Transcription
Curso de normas de Buena PrácticaClínica (BPC) para investigadores ycolaboradores de ensayos clínicos.1ª ediciónAspectos prácticos en la realización de losensayos clínicos:Cuaderno de recogida de datos,Monitorización y Archivo.17 de mayo de 2011, Hospital Universitari Vall d’HebronSilvia García MatasGestora de proyectos y monitoraUnitat Central d’Investigació Clínica i Assaigs Clínics (UCICAC)Vall Hebron Institut de Recerca (VHIR)sgarcia@ir.vhebron.netucicac@ir.vhebron.net
Temas a tratar1.Monitorización1.1. Definición y Objetivos1.2. El Monitor1.3. Monitor vs Enfermera del estudio o data manager1.4. Responsabilidades del monitor1.5. Tipos de vistas1.6. Deficiencias encontradas en las visitas de monitorización2. Archivo2.1. Definiciones2.2. Normativa2.3. Documentación necesaria para un ensayo2.4. Resumen documentos esenciales del ensayo a archivar3. CRD3.1. Definición y responsabilidades3.2. Requisitos y finalidades3.3. Anotaciones en el CRD papel3.4. Ejemplos de CRD electrónicos
1. Monitorización1.1 Definición y ObjetivosDefinición:Monitorización: Acto de vigilar el progreso de un ensayo clínico y de asegurarque es realizado, registrado y comunicado de acuerdo con el protocolo,los procedimientos normalizados de trabajo (PNT), la buena práctica clínica(BPC) y los requisitos reguladores aplicables.Objetivos:La regulación de la FDA (21 CFR 312.56) y las ICH Guidelines ICH-E6 5.18identifican tres razones para la realización de la monitorización: Velar por la seguridad de los pacientes y que se respeten su derechos éticos. Comprobar que los datos recogidos durante el estudio son verificables ( no selos ha inventado nadie) , exactos (no hay error al transcribirlos al cuaderno derecogida de datos) y completos. Asegurar que el ensayo se lleva a cabo según lo establecido en el Protocolo,según las Normas de Buena Práctica Clínica y la legislación española (RealDecreto 223/2004)
1. Monitorización1.2 El monitorMonitor / CRA (Clinical Research Associate)Artículo 36. REAL DECRETO 223/2004 de 6 de febrero de 2004.Profesional capacitado con la necesaria competencia clínica, elegido por elpromotor, que se encarga del seguimiento directo de la realización del ensayo.Sirve de vínculo entre el promotor y el investigador principal, cuando éstos noconcurran en la misma persona.Requisitos de un monitor Ser nombrados por el promotor y actuar como la pral. vía de comunicación entreel promotor y el investigador para discutir la evolución del estudio. Estar debidamente formados y tener el conocimiento científico y/o clíniconecesario para monitorizar el ensayo adecuadamente. La cualificación del monitorestará documentada (licenciado ciencias de la salud, alto nivel inglés,conocimientos informática, diploma/master en EECC). Familiarizados con el medicamento en investigación, el protocolo, la hoja deinformación escrita del consentimiento informado y cualquier otra informaciónescrita que sea facilitada a los sujetos, los PNT del promotor, la BPC y lalegislación vigente.
1. Monitorización1.2 El monitor¿Cómo reconocer a un monitor?
1. Monitorización1.2 El monitorEl promotor de un estudio clínico está obligado por ley a monitorizar losensayos en activo.¿Quién puede ser monitor?CRO (Organización de investigación por contrato / Contract ResearchOrganization) , Persona u Organización (comercial /académica u otra)contratados por el promotor para realizar una o mas funciones o deberesdel promotor en relación con el ensayoEl monitor no debe formar parte del equipo investigador.
1. Monitorización1.3 Monitor vs Enfermera del estudio o data manager
1. Monitorización1.4 Responsabilidades del monitorResponsabilidades del monitor:Real Decreto 223/2004 Artículo 36. MonitorReclutamiento de los pacientes Asegurar la correcta obtención del CI antes deincluir el paciente en el ensayo . Verificar que los pacientes incluidos cumplen loscriterios de selección del protocolo. Notificar el ritmo de reclutamiento.Gestión de actividades (opcional) Solicitar autorizaciones al CEIC y AEMPS Gestionar contratos Garantizar la comunicación entre investigador ypromotor Garantizar que el investigador recibe istros) Comprobar que el almacenamiento, distribución,devolución, destrucción y documento de losmedicamentos en investigación es seguro yadecuado.Cumplimiento del protocolo Visitar al investigador antes, durante ydespués del ensayo para verificar que cumplecon el protocolo: Cerciorarse que los investigadores y centrodonde se realizará el ensayo son adecuados. Explicar el protocolo a l equipo investigador. Verificar los datos originales. Notificar los Acontecimientos Adversos Detectar e informar al promotor de lasdesviaciones. Remitir al promotor informes de la visitas demonitorización y elaborar informes demarcha del ensayo.Gestión de registros Determinar si el investigador mantiene losdocumentos esenciales. Verificar la exactitud e integridad de los datosincluidos en el CRD vs documentos fuente. Resolver con los investigadores las queries(incongruencias en los datos recogidos).
1. Monitorización1.4 Responsabilidades del monitor¿Qué quiere un monitor?
La cruda realidad .
1. Monitorización1.5 Tipos de vistasTipos de visitas de monitorización durante un ensayo clínico1. Visita de selección2. Visita de inicio3. Visita de monitorización4. Visita de cierreVisitas de monitorizaciónVisita deselecciónVisita deinicioTras inclusión 1erpacienteTras últimavisita últimopacienteQueries ycierre Base deDatosVisita decierreInforme final ypublicación
1.5 Monitorización: Tipos de vistasa) Visita de selección del equipo investigador y centroVisitas de monitorizaciónVisita deselecciónVisita deinicioTras inclusión1er pacienteQueries y cierre Base de DatosTras última Visita de cierrevisita últimopacienteInforme final ypublicaciónLa selección de investigadores y centros posiblemente es el 50% el éxito deun ensayo clínico el otro 50% es el resto (fármaco, protocolo, áreaterapéutica, oportunidad, CRO, Monitor).¿Quién debe seleccionar a investigadores y centros? El promotor o la CRO.Aspectos a tener en cuenta: Tener Experiencia previa del personal siguiendo Buenas Practicas Clínicas Tener el equipo y el personal adecuado para el ensayo clínico (experiencia ycualificación en investigación en el área terapéutica del EC) Tener tiempo para realizar las actividades del ensayo Cantidad y disponibilidad de pacientes de acuerdo con los criterios de selecciónestablecidos en el protocolo. Capacidad para cumplir plazos El interés en participar.Si el promotor considera idóneo con esta información al investigador y éste quiereparticipar. Se envía al investigador un acuerdo de confidencialidad y se le envía elProtocolo, Manual del Investigador, Memoria económica, etc.
1.5 Monitorización: Tipos de vistasa) Visita de selección del equipo investigador y centro
1.5 Monitorización: Tipos de vistasa) Visita de selección del equipo investigador y centro
1. 5 Monitorización: Tipos de vistasb) Visita de inicioVisitas de monitorizaciónVisita deselecciónVisita deinicioTras inclusión1er pacienteQueries y cierre Base de DatosTras última Visita de cierrevisita últimopacienteInforme final ypublicaciónDespués de obtener la autorización del CEIC, AEMPS. Tener la conformidad delcentro y el contrato entre el promotor y centro firmado. Antes de la inclusión delprimer paciente. Normalmente antes de la llegada de la medicación al centro.Formación de todo el equipo investigador que ejecutará el EC y se visitarántodos los servicios implicados. Propósito y motivo del EC Aspectos prácticos de la medicación de estudio Buenas Prácticas Clínicas y normativa aplicable Protocolo: criterios inclusión/ exclusión,reclutamiento pacientes, retirada pacientes,procedimiento y visitas Hoja de Información al Paciente yConsentimiento Informado (CI) Cuaderno de Recogida de Datos Acontecimientos Adversos comunesdescritos en el manual del investigador Notificación de AA Mantenimiento archivo de investigador Procedimientos laboratorio Verificación de datos y acceso a losdocumentos fuente Visitas de monitorización (frecuencia) Posibles auditorias
1. 5 Monitorización: Tipos de vistasc) Visitas de monitorizaciónVisitas de monitorizaciónVisita deselecciónVisita deinicioTras inclusión1er pacienteQueries y cierre Base de DatosTras última Visita de cierrevisita últimopacienteInforme final ypublicación Confirma que los investigadores, el equipo, las instalaciones y materialsigue siendo adecuado. Confirma que el ensayo se realiza según el protocolo, según lanormativa vigente, según BPC y notificando correctamente losAcontecimientos Adversos en especial los Graves e Inesperados. Revisa de los Consentimientos informados firmados Verifica la correcta recogida de los datos de los documentos fuente enel Cuaderno de recogida de datos (CRD). Recoge hojas del CRD en papel o validar datos en el eCRD. Comprueba el correcto manejo de la documentación de estudio. Comprueba que el archivo está actualizado.
1. 5 Monitorización: Tipos de vistasc) Queries después de las visitas de monitorizaciónVisitas de monitorizaciónVisita deselecciónVisita deinicioTras inclusión1er pacienteQueries y cierre Base de DatosTras última Visita de cierrevisita últimopacienteInforme final ypublicaciónEl monitor revisa los datos del Cuaderno de Recogida de Datos: puede serque sea necesaria alguna aclaración o verificación posterior de los datos.Genera “data query” y se envían al investigador.El investigador responde, revisa la base de datos y modifica el dato si fueranecesario.En todo este proceso es necesario la trazabilidad de los datos.
1. 5 Monitorización: Tipos de vistasd) Visita de cierreVisitas de monitorizaciónVisita deselecciónVisita deinicioTras inclusión1er pacienteTras últimavisita últimopacienteQueries y cierre Base de DatosVisita de cierreInforme final ypublicaciónMotivos para visita de cierre: Fin de estudio. No cumplir con el protocolo No cumplir con las BPCs A petición del investigador Bajo reclutamiento¿Quién? La realiza el coordinador del estudio y/o monitor. Debe estar elInvestigador Principal y colaboradores.¿Cuándo? Tras última evaluación del último paciente y cuando estén resueltastodas las queries.Objetivos: Archivar la documentación del ensayo Recogida de toda la medicación y material sobrante. Archivo del centro revisado y bien almacenado. Notificación final del ensayo (CEICs, AEMPS)
1. Monitorización:1.6 Ejemplos de deficiencias encontradas en las visitasEjemplo de deficiencias más comunes durante las visitasCI incompleto no seha completado esteapartadoEl nombre del Dr.no lo ha escrito elpacienteFalta el segundoapellidoEl Investigador haescrito las fechas ylos nombres . Teníaque haberlos escritoel eic/documentacionEnsayoCli.htm
1. Monitorización:1.6 Deficiencias encontradas en las visitas de monitorizaciónFaltacompletar elCI con larelación delrepresentanteFalta segundoapellido delrepresentantelegal y delpacienteEl investigadorha completadolos datos enlugar delrepresentanteFalta la firmay escribir elsegún doapellido delInvestigdorEl investigadorno ha escritosu segundoapellidoFalta escribir elsegúndo apellidodel representantelegalSe ha utilizadoun sello parafechar el CI
1. Monitorización:1.6 Ejemplos de deficiencias encontradas en las visitasEl testigo no ha completado elnombre y los dos apellidos delinvestigadorEl investigador hacompeletado yfechado el CI
1. Monitorización:1.6. Deficiencias encontradas en las visitas de monitorizaciónDeficiencias más comunes de las visitas demonitorización Consentimiento Informado (CI) obtenido después de haberrealizado algún procedimiento para el ensayo clínico Reconsentimientos no firmados o firmados muy tarde. CI no completado enteramente por el paciente; fecha no escritapor el paciente, nombre del paciente escrito por el Investigador Información insuficiente en la historia sobre la firma del CI; no seespecifica si es el consentimiento general o los opcionales porsubestudios o reconsentimientos. Proceso de obtención del CI inadecuado. Firma de una versión de CI obsoleta.
1. Monitorización:1.6. Deficiencias encontradas en Documentos fuente y CRDDeficiencias encontradas en documentos fuente y CRD Discrepancias entre los documentos fuente y los CRD. AAs y medicaciones concomitantes no registrados o registradosinadecuadamente (faltan dosis, fechas de inicio y de fin, etc ). Ausencia de documentos fuente para algunos datos(eligibilidad, fechas, cumplimientos, etc ) Datos anotados en la historia de forma retrospectiva. Tachones y uso de líquido corrector Entradas en la historia clínica no identificadas. Analíticas y ECG no firmados. Retraso en la entrada de datos o entrada incompleta de datos. Retraso en la resolución de discrepancias (queries).
1. Monitorización:1.6. Deficiencias encontradas en la Adherencia al protocoloDeficiencias en la adherencia al protocolo: Pacientes que no cumplen los criterios de inclusión-exclusióndel ensayo. Pruebas de selección fuera de fecha según el calendariode actividades. Pruebas de selección no realizadas. Aleatorización de un paciente sin conocer el resultado deuna prueba. Desviaciones del protocolo. Valores de analíticas no realizados. Visitas o pruebas hechas fuera del calendario de actividades. Cuestionarios de calidad de vida no hechos. ECGs, constantes vitales, datos demográficos no hechos. Medicación concomitante prohibida tomada por el paciente.
1. Monitorización:1.6. Deficiencias encontradas en Gestión de acontecimientos adversosDeficiencias en la Gestión de los acontecimientosadversos: Acontecimientos Adversos (AA) no registrados en el CRD Documentación insuficiente sobre los AAs en losdocumentos fuente (causalidad, intensidad, fechas inicio/fin, etc.). Acontecimientos Adversos Graves (AAGs) notificados tarde, fueradel periodo exigido (el investigador principal tiene como máximo24h desde que se conoce el caso para informar al promotor).Notificaciones de AAGs deficientes (poca información) AAGs no notificados.
1. Monitorización:1.6. Deficiencias encontradas en la Mediación del estudioDeficiencias encontradas en la medicación del estudio: Registros ausentes o incompletos de la medicacióndispensación/prescripciones Contabilidad no realizada o medicación no devuelta. Errores en la administración del fármaco (pauta incorrecta) Valoración del cumplimiento del paciente. ¿Quién lo valora? Condiciones de conservación del fármaco inadecuadas (no accesolimitado, control deficiente de la temperatura, etc.) Investigador sin acceso a poder abrir el ciego en unestudio con un IVRS o con sobres sellados. ¿Qué procedimientodispone el centro para localizar al IP para abrir un ciego en caso denecesidad?
1. Monitorización:1.6. Deficiencias encontradas en Instalaciones y personal del estudioDeficiencias en las instalaciones y personal del estudio Información contenida en el listado de delegación detareas incompleta o inexacta. El Investigador Principal (IP) es elresponsable del listado y de las tareas asignadas. Comunicación tardía de la implicación de nuevos Investigadorescolaboradores. Insuficiente supervisión por parte del IP tanto del ensayocomo de las tareas delegadas a sus colaboradores. No registros de formación específica del equipo investigador en elprotocolo/enmiendas o en BPCs (Minutas sesiones clínicas). Falta de comunicación entre los servicios implicados en el Ensayo. Falta de certificaciones de calidad de los aparatos/calibraciones.
2. Archivo2.1. DefinicionesDocumentos esenciales: aquellos documentos que de formaindividual y colectivamente permiten la evaluación de la realización delensayo clínico y la calidad de los datos obtenidos. Sirven para demostrarel cumplimiento con los estándares de BPC y de los requisitosregulatorios del Investigador, Promotor y Monitor.El archivo es donde se guarda toda la documetación referente a de unensayo clínico que nos servirá para demostrar que se ha realizado segúnla legislación vigente y el protocolo.Su correcto mantenimiento (durante el desarrollo del estudio) permite unbuen manejo del mismo por parte de Investigador, Promotor y Monitor.“Lo no documentado no ha ocurrido”
2. Archivo2.2. NormativasExisten normativas que regulan, documentos a conservar y tiempo de retención.InvestigadorDirectiva Europea 91/507/EEC:La identificación de los sujetosConservar hasta 15 años tras lafinalización del ensayo clínicoHistorias clínicas, datos analíticos etc.Tanto como sea posible.PromotorDocumentos esenciales:Conservar hasta 15 años tras lafinalización del ensayo clínico
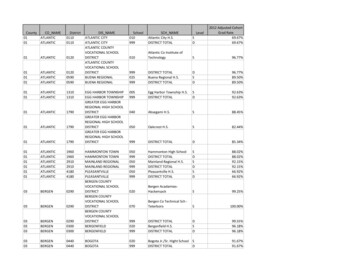
2. Archivo2.3. Documentación necesaria para un ensayoSegún BPC la documentación necesaria para un ensayo clínico es la siguiente:
2. Archivo2.3. Documentación necesaria para un ensayo
2. Archivo2.3. Documentación necesaria para un ensayoEnsayo clínico Burocracia
2. Archivo2.4. Resumen documentos esenciales del ensayo a archivarVisitas de monitorizaciónVisita deselecciónQueries y cierre Base de DatosVisita de Tras inclusión Tras última Visitainicio 1er paciente visita último de cierrepacienteAntes del inicio del ensayo Manual de informaciónbásica del producto Protocolo del estudio Cuadernos de recogidade datos Muestras del producto ycódigos de randomizaciónDurante el estudio Información no publicada quepueda interferir con el desarrollodel estudio Actualizaciones del manual deinformación del productoInforme final ypublicaciónDespués de finalizarel estudio Informe final
3. Cuaderno de Recogida de Datos (CRD)3.1. Definición y responsabilidadesLas normas de BPC obligan a guardar todos los datos de forma quese pueda reconstruir un estudio.CRD: Cuaderno de Recogida de Datos CRF Case Report FormDefinción:Documento impreso, óptico o electrónico, diseñado para recoger todala información requerida por el protocolo para cada paciente incluidoen el ensayo.Tratamiento del CRD y responsabilidades:Promotordiseño , suministro del CRD y validación delprogramas de tratamiento de datosInvestigadorcumplimentación correctaMonitorrevisión y seguimiento del CRDData Managementtranscripción de los datos a la Base de Datos
3. Cuaderno de Recogida de Datos3.2. Requisitos y finalidadRequisitos del CRD Simplicidad Evitar ambigüedad. Exigir lo mínimo imprescindible. Reflejar el diseño del Ensayo Clínico. Reflejar claramente los objetivos y variables del Protocolo. Utilidad práctica. Personalizado. Facilitar entrada de datos y tratamiento estadístico. Seguir las Buenas Prácticas Clínicas de manera que:Permita la verificación de datos.Los Datos escritos deben ser exactos y legibles.Los datos que refleja deben ser coherentes con losdocumentos fuente.Las correcciones deben estar validadas.Debe aparecer las omisiones, retiradas, los AA,enfermedades y tratamientos concomitantes.Finalidad del CRDEl CRD sirve para verificar la calidad y exactitud de los datos obtenidos y lafiabilidad de los datos originales. Los datos no recogidos en el CRD nopueden aparecer en el resultado final del estudio.
3. Cuaderno de Recogida de Datos3.3. Anotaciones en el CRD en papelAnotaciones en el CRD en papel Todas las entradas de datos serán fechadas y firmadas por elinvestigador. Usar papel de calidad. No se puede utilizar ni lapiz ni líquido corrector (TIPP-ex). Si el dato primario proviene de un aparato que usa papel térmico,fotocopiar indicando que es una copia del original : firmar y fechar.Las modificaciones deben ser: Tachando con una linea fina el dato anterior que debe poder leerse. Poner el dato correcto al lado. Indicar a pie de página la causa de la rectificación firmar y fechar.
3. Cuaderno de Recogida de Datos3.3. Anotaciones en el CRD en papelCódigo del EC no comercialCódigo del EC no comercialCódigo de ECCódigo de EC
3. Cuaderno de Recogida de Datos3.3. Anotaciones en el CRD en papelCódigo del Ensayo clínico no comercialCódigo del Ensayo clínico
3. Cuaderno de Recogida de Datos3.4. Ejemplos CRD electrónicos
3. Cuaderno de Recogida de Datos3.4. Ejemplos CRD electrónicos
3. Cuaderno de Recogida de Datos3.4. Ejemplos CRD electrónicos
3. Cuaderno de Recogida de Datos3.4. Ejemplos CRD electrónicos
3. Cuaderno de Recogida de Datos3.4. Ejemplos CRD electrónicos
Ideas principales La monitorización de un ensayo clínico es necesaria y esta definidalegalmente. El monitor es el interlocutor entre el investigador y el promotor. Función primordial del monitor es velar por la seguridad y derechos éticos delos pacientes, comprobar que los datos recogidos durante el estudio sonverificables, exactos y completos, asegurando que el ensayo se lleva a cabosegún lo establecido en el Protocolo, según BPC y la legislación española. Los documentos esenciales en un ensayo clínicos son autorización del CEICy AEMPS, protocolo, Hoja Información al Paciente y Consentimiento Informado,CRD, Manual del Investigador o ficha técnica, seguro, informes demonitorización, informes de seguimiento y seguridad del ensayo. El monitor corrobora que la información principal de un ensayo está bienrecogida en el CRD. Los datos que no están recogidos en el cuaderno de recogida de datos nopueden formar parte de los resultados del ensayo.
Unitat Central d’Investigació Clínica i Assaigs Clínics (UCICAC):Las personasB. RipollI.Fuentes S.Garcia R.SimóE. López A.Salgado E.Carceller C.Cases P. Fernández M.AngladaMUCHAS GRACIAS !Consultas a:ucicac@ir.vhebron.netC. CasesA. Salgado S. Pérez-Hoyos E. LópezX. Vidal
1. 5 Monitorización: Tipos de vistas. Motivos para visita de cierre: Fin de estudio. No cumplir con el protocolo No cumplir con las BPCs A petición del investigador Bajo reclutamiento ¿Quién? La realiza el coordinador del estudio y/o monitor. Debe estar el