Transcription
Application Spotlight: AgrigenomicsSequence-Based Genotyping BringsAgrigenomics to a CrossroadsFor some applications, sequence-based genotyping provides a lower-costalternative to microarrays in performing genetic variation studies.IntroductionToday, agrigenomics researchers have a wide varietyof technologies at their disposal for collecting geneticinformation. Array-based approaches to SNP screeninghave been the method of choice in analyzing andassociating traits with regions of the genome for manyplants and animals. As sequencing costs continue todecline, new approaches that leverage next-generationsequencing (NGS) technology are being developed toperform genotyping studies. The term next-generationsequencing–based genotyping (NGG), orsequence-based genotyping, encompasses genotypingmethods that leverage NGS technology. NGG includestargeted, reduced representation, andhybridization-based approaches to discover andgenotype SNPs, often simultaneously in manyindividuals or specimens. This application spotlightprovides insight into different NGG methods, theirbenefits, and the role that conventional array technologywill play in the future.Arrays Pave the Way in AgrigenomicGenotypingIn the late 1980s, researchers began identifying specificregions of DNA that influenced phenotypic traits incertain species. Efforts soon turned to the developmentof accurate and cost-effective genetic tests that couldcharacterize the genotype of these regions in a sample.User-friendly PCR-based markers such as short tandemrepeats (STRs or SSRs) were ultimately replaced withsingle nucleotide polymorphisms (SNPs) as the chosenmarker for genotyping studies. Not only are SNPspresent in high abundance within genomes, but whenscreened in high densities for a given species, theyenable the efficient tracking of the transfer of geneticregions from parent to offspring. SNP-based assays arenow routinely used to identify trait–marker associationsand perform genomic selection, parentage testing, andmarker-assisted selection.1Optimizing marker density to detect trait associations isone of the main challenges when developing genotypingtools. Trait associations rely on detecting recombinationunits (haplotype blocks), making it essential to optimizemarker density for the targeted “diversity population”for genotyping to be performed at an affordable costper sample.Many critical steps are involved in building robustgenotyping arrays, including initial SNP discovery,diversity assessment, and SNP selection.2–3 After thesesteps, a filtered, high-quality subset of SNPs is deployedonto a high-density genotyping platform, such as theInfinium Assay. The cost per sample often limits the useof SNP microarrays to research applications where thescreening populations are small.Yet, many agricultural applications could benefitprofoundly from genotyping, including the screening ofbreeding populations.4 By leveraging genetic screening,farmers and livestock breeders could gain immediatefeedback, supporting better informed breedingdecisions and accelerating their return on investment(ROI). Genotyping tools with a lower cost per samplecould enable genetic screening to be performedroutinely on large populations, with an attractive ROIoffsetting the implementation cost of the technology.
Application Spotlight: AgrigenomicsSequencing Advances Can Deliver MoreCost-Effective GenotypingThe rapid evolution of sequencing technology hasresulted in higher throughput and a lower cost persample, often positioning NGG as a cost-effective andefficient agrigenomics tool for genotype screening,genetic mapping, purity testing, screening backcrosslines, constructing haplotype maps, and performingassociation mapping and genomic selection.5–7 Thenumber of NGG methods continues to grow, with eachoffering the fundamental benefits sequencing provides,including reduced ascertainment bias, identification ofvariants other than SNPs (small insertions, deletions,and microsatellites), and an ability to performcomparative analysis across samples in theabsence of a reference genome (Table 1).Methods of Sequence-BasedGenotypingFor small genomes (eg, Drosophila) or high-profileresearch species (eg, Arabidopsis), genotyping andvariant screening can be completed using standardwhole-genome sequencing/resequencing (WGS)methods relative to a reference. For larger genomeswhere funding is limited, sequence-based genotyping(or NGG) methods have been developed.NGG advances are greatest for methods that can beperformed at a lower cost than WGS. Crop researcherssupporting applications in genomics-assisted breedingand genomic selection have been the primary drivers ofdeveloping lower-cost protocols.8–9Table 1: Genotyping by Sequencing Benefits and Considerations.Benefits OverArray GenotypingExplanationConsiderationsNGG methods often use homebrew librarypreparation with multiplexing.Per-sample costs 20 USD. Populations with low diversity (eg, cotton) will exhibit fewerpolymorphisms than those with higher diversity (eg, maize).Therefore, cost per data point will be higher for low-diversityspecies.10 Targeted methods of enrichment and restriction enzyme methodsboth require fine-tuning of coverage across highly multiplexedsamples for optimal cost benefit.NGG methods are well-positioned toleverage future sequencingcost improvements. As sequencing protocols and analyses are defined, published, andshared, consistency in data management and sample and genebank tracking will be key to optimizing resources.11 As protocols use higher levels of sample multiplexing and lowercoverage per individual (eg, skim sequencing), tolerance forambiguity in heterozygote detection must be considered. Sequencing data analysis methods, while constantly improving,are still less streamlined than array methods for data analysis. Thiscan be a barrier for new users with species that have little geneticinformation (ie, no reference genome).Low ascertainment biasAscertainment bias, especially inhigh-diversity species, presents challengeswith array genotyping for lines that havea parental background different from thereference or SNP discovery population.Sequencing methods have a lower burdenof a priori knowledge. Pulldown or amplicon methods have potential to have some bias ifthey depend on hybridization. Restriction site–associated methodswill be bias free if restriction sites are conserved among targetedlines of interest.5,9Increased dynamic rangedetection offered bysequencing inpolyploid speciesHigher allele dosage detection levels ofsequencing over array methods enableincreased allele detection sensitivity ofmultiple genomes in polyploid species. Filtering criteria for sequencing data might require adjustments foreach species protocol defined. Illumina GenomeStudio software supports automatedpolyploidy calling.Some sequencing protocols, like thoserelying on restriction enzyme cut sites, canbe completed in the absence of areference genome.5 Transcriptome assembly or contigs (eg, 10 kb) can act as aputative reference from some sequencing applications. When using a reference that is distant from the targeted species(ie, use of the bovine as a reference for whale), there is some riskthat a high mismatch rate for rare variants might bias toward highMAF SNPs.Low-cost genotyping nowLower-cost genotyping inthe futureInsight into non-modelgenomes where no a priorigenomic informationis available
Application Spotlight: AgrigenomicsSkim SequencingHybridization-Based MethodsLow coverage or scalable/tunable skim sequencinghas been demonstrated in wheat chromosomal linesas effective for SNP discovery and is useful for detaileddiversity analysis, marker-assisted selection, andsequence-based genotyping.12–13 It offers numerousadvantages, including an established library preparationprotocol, an established informatics application pipelinethat enables SNP calling within reads rather than relativeto a reference, and redundancy checks that minimizefalse positives.12 The amount of data generated usingskim sequencing can also be modulated byrerunning the samples to increase coverage, avoidinglibrary preparation optimization and challenges insample tracking.Hybridization-based approaches include solidsubstrate as well as liquid hybridization methods,using oligonucleotide specificity to bind to and isolatecomplementary sequences. To leverage sequencingcapacity and optimize costs, these methods rely onmultiplexing samples enriched using the same probesets. Solid phase hybridization is completed afterlibrary preparation, where regions of the genomethat hybridize are retained and those that do not arewashed away. The more common methods of solutionhybridization typically take advantage of biotinylatedprobes or RNA baits to facilitate capture of targets.Hybridization capture has an advantage in genotypingallotetraploids because it can enable homologousgenomes to be differentiated.17EnrichmentBy using PCR or hybridization probes, a suite ofmethods can be used to isolate a specific genomicfraction by either removing unwanted components(target enrichment, Figure 1A) or selecting desiredtargets (targeted pulldown, Figure 1B) for subsequentsequencing.14–15 Sequencing is focused on regionsof interest, offering sufficient overlap in sequencingcoverage to call SNPs reliably. Particularly in plants,these methods avoid lost sequencing space toduplicated or otherwise undesirable areas ofthe genome.14PCR-Based MethodsMany PCR-based genotyping methods have beendeveloped. They include direct sequencing of PCRamplicons, long-range PCR sequencing wherefragments are sheared in library preparation, and the useof molecular inversion probes to target long regions thatare circularized with a ligase before amplification. Thesemethods can pose challenges in scaling marker andsample multiplexing (multiple samples per flow cell orlane) to leverage NGS throughput and minimize costs.Challenges include accurately optimizing multiplexreaction conditions to capture all targeted regionsuniformly.14,16 There are several commercial-based PCRmethods that facilitate optimal multiplex conditions,including Illumina TruSeq Custom Amplicon.Targeted EnrichmentTargeted enrichment approaches are ideal for pristinegenomes (eg, bovine, rice) where there is a prioriknowledge for regions of interest, such as markers forloss of function or trait associations used inmarker-assisted selection. They are powerful methodsfor SNP discovery and fine mapping of recombinationbreakpoints. For example, researchers studying wheatused a sequence capture assay for targetedresequencing of a 2.2 Mb exon region and identified4,000 SNPs and 129 indels suitable for differentiatingbetween cultivated and wild wheat populations.17A need to reduce costs has been the primary driverof the evolution in sequence-based approaches togenotyping. Therefore, cost-effective targeted andenrichment methods will be increasingly important toallow researchers to choose their markers of interestas more genomes are assembled and referenced.Restriction Enzyme Methods: RE-GBS,RAD-Seq, and ddRADSeqThe biggest advances in NGG affordability havebeen achieved using restriction enzyme methods ofreducing the representation of the library for subsequentsequencing. Restriction enzyme GBS (RE-GBS),restriction site-associated sequencing (RAD-Seq),and ddRADSeq methods use restriction enzymes togenerate fragments for sequencing. They provide areduced, genome-wide representation with data that
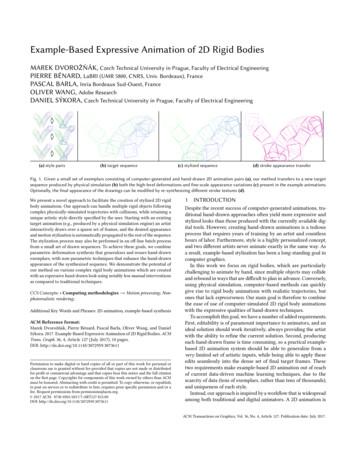
Application Spotlight: AgrigenomicsA. Amplicon-Based Targeted SequencingPCR1 reaction 1 amplicon/sampleMultiplexed PCR1 sample/reaction 60 amplicons/sampleCombined, indexed, multiplexed PCR96 samples/reaction 180 ngB. Hybridization-Based Enrichment SequencingLigate adaptersPool andhybridize target probesIsolated fragmentscorresponding to targeted regionsSequenceGene ModelMapped ReadsC. Restriction Enzyme Reduced Representation Sequence-Based GenotypingRE digest and ligate adaptersPoolSize select (optional)SequenceRestriction enzyme cut siteGenomic DNAReducedRepresentationFigure 1: NGG Methods for Discovering and Genotyping SNPs. Amplicon-based targeted resequencing methods (panel A) adapted fromMamanova et al, 201016 and Liu et al, 2012.18 Hybridization-based enrichment sequencing methods (panel B) adapted from Mamanova etal, 201016 and Cronn et al, 2012.10 Restriction enzyme reduced representation sequence-based genotyping methods (panel C) adapted fromAndolfatto et al, 2011.19can be aligned, compared, and screened for SNPvariants (Figure 1C).5,8,9,20 NGS-compatible fragmentlibraries enable massively parallel and multiplexedsample sequencing, facilitating the rapid discovery andgenotyping of tens to hundreds of thousands of SNPsacross large populations.RE-GBS protocols, initially established for crops likemaize and wheat, have advantages in cost per sampleand application in species where there is no a prioriknowledge of the genome. The application of RE-GBSis especially powerful in mapping populations, or closelyrelated groups of samples, such as candidates for
Application Spotlight: Agrigenomicsgenomic selection. If populations are more divergentthan expected or target novel species, RE-GBSprotocols can require optimization (beyond publishedprotocols) to customize coverage and minimize missingdata. For example, high divergence across targetedsamples can result in missing data, complicatingdownstream analysis, whereas low divergence canresult in a lower number of detected SNPs.The advantages of RE-GBS are many, making theprotocol development for species-specific applicationsrewarding.8 Reduced ascertainment bias overarray-based methods, the ability to discover andcharacterize polymorphisms simultaneously, and thegeneration of valuable genetic information for a low( 20 USD) cost per sample (excluding bioinformatics)make this a method of choice for those moving fromarray methods to genotyping by sequencing. RE-GBSdata analysis methods are supported with open-sourceanalysis tools (eg, TASSEL) that can be tailored for cropsof interest using a command-line interface. Table 2(reproduced from Nielsen et al, 201121) shows a listof available non-commercial NGS genotyping callingsoftware. Nielsen et al also present a workflowfor converting NGS data into SNP calls (Figure 2).RAD-Seq protocol enhancements have been primarilyfocused on increasing the level of multiplexing to reducecost and eliminate expensive steps in the protocolworkflow, such as random shearing and the subsequentneed for end repair. Examples of methods that eliminaterandom shearing include MSG,19 CRoPS,22 andddRADSeq.20 The ddRADSeq method has been usedto refine size selection, recovering a “tunable number ofregions” distributed randomly throughout the genomeat a reported library preparation cost of 5 USD persample and input amounts as low as 100 ng of startingDNA.20 This approach also implements a two-indexcombinatorial multiplex system (n*m individuals usingn m indexes), a sequence filter analysis toolkit, anda sample tracking data management tool availablethrough a Google Docs interface. High-throughputdata management and sample tracking are critical forimplementing any sample screening method in breedingand germplasm tracking.11Table 3 summarizes published sequence-basedgenotyping methods, including PCR-based,hybridization-based, and restriction enzyme approaches.Image analysis and base callingRead mappingRealign, remove duplicate reads, and recalibrate quality scoresMulti-sample callingSingle-sample callingPromote candidate SNPset and genotype callsusing non-linkage-based,multi-sample analysisRefine candidate SNPset and genotype callingusing linkage-based analysisIdentify SNPs andassociated genotypes usingsingle-sample analysisSNP filtering and SNP or genotype quality score recalibrationFigure 2: Converting NGS Data Into Genotype Calls.Reproduced from Nielsen et al, 2011.21 First, pre-processing stepstransform NGS data into aligned reads with quality scores that indicateconfidence. Next, SNP or genotype calls are made using amulti-sample or single-sample calling procedure, depending on thenumber of samples and depth of coverage. Finally, post-processingsteps filter the called SNPs.
Application Spotlight: AgrigenomicsTable 2: Available Non-Commercial NGS Genotype-Calling Software.SoftwareAvailable FromCalling .htmlSingle-sampleHigh-quality variant database (eg, -sampleAligned ligned igned beagle.htmlMulti-sample LDCandidate SNPs, genotype likelihoodsmathgen.stats.ox.ac.uk/impute/impute v2.htmlMulti-sample LDCandidate SNPs, genotype rd/QCALLMulti-sample LD‘Feasible’ genealogies at a dense set of loci,genotype lti-sample LDGenotype likelihoodsAdapted from Nielsen et al, 2011.21Determining Sequence DepthHigh-throughput microarrays (millions of SNPs forthousands of samples) have been used for yearsto perform genotype screening, with heterozygotedetection exceeding 99.99% through optimizedprobe design. For the detection of heterozygosity,NGG methods depend upon sequencing depth, withincreased depth resulting in increased cost per sample.When the goal is to detect parental lines that are fixedfor alternate alleles, heterozygotes are infrequent and oflittle consequence. As a result, multiplexing can be highand coverage per sample as low as 1 to meet projectgoals. For applications requiring heterozygote detection,missing or ambiguous genotypes can be overcome byresequencing the library in greater depth or by using“soft” bin assignment informatics approaches19 thatfacilitate imputation to missing allele states. Li et al,2011 provide useful modeling analysis for depth ofcoverage needed to detect SNPs with certainty anddetect heterozygotes from sequencing runs with 2 ,4 , 6 , and 30 coverage and a range of minor allelefrequencies in the population.23 Tolerance for missingdata can be a critical consideration for sequence-basedgenotyping decisions.Independent of the NGG method chosen, there aretradeoffs among factors of marker density, sequencedepth, and degree of multiplexing for cost per sample.In RE methods, the more markers targeted (eg, 4-baseover a 6-base enzyme cutter), the more fragmentsare created and the more sequencing is required.Improvements can be expected to lower costs ofsequencing with longer reads and evenness ofcoverage among multiplexed individuals. All of thesewill allow for quicker associations between genomicregions and traits at a lower cost per sample andimproved implementation of marker-assisted breedingin agriculture species.The Value of ArraysWhile they are no longer the only solution, arraymethods are often still an excellent fit for screeningapplications, especially with well-annotated genomeswhere established trait associations and loss-of-functionvariants are known. For example, many agricultureresearch communities need tools for routine testingof known markers with consistent high-throughputdata analysis, where volume pricing offers a cost persample that tips the scale toward an array approachover an NGG approach. When whole communitiesconverge on a common tool, there is an opportunity toleverage the diverse data sets and develop downstreammethods for imputation and proprietary custom orAdd On content. For example, lower-density arrays( 50,000 SNPs indels) are useful as base contentfor building proprietary Add On beadpools that seedcompanies can use to build a proprietary array withpublic and private marker content combined in asingle chip. The combination of array- and sequencebased genotyping approaches has already contributedsignificant value to the dairy cattle breeding industry.As shown by the 1,000 Bull Genomes Project, mergingthe 2 technologies allows for highly accurate imputationfor related individuals in a combined data set.24 Illuminaoffers comprehensive sequencing and array solutionsthat can be tailored to any species.
Application Spotlight: AgrigenomicsTable 3: Published Sequence-Based Genotyping Methods.MethodType of MethodDescriptionPCR basedOften used in metagenomics applications where 16S fragments are targeted. Labor intensiveto amplify and tag multiple targets to optimize sequencing coverage. Difficult to scale currentlyto leverage sequencing output to drive down price per sample.PCR basedLong-range PCR ( 35 kbp, typically 3–10 kbp) can be used to target regions that then requireshearing before library preparation. Challenges include equimolar pooling of sample/fragments.Tends to be a drop in coverage at the ends that can be resolved by increasing ampliconoverlap to a minimum of 100 bp.10,16Whole genomeMolecular inversion probes, single-stranded oligonucleotides with common linker flanked bytarget-specific sequences, anneal to target sequence and become circularized by a ligase.PCR amplification and products are sequenced directly. Suited for few targets and high samplenumbers ( 100 samples).25–27Whole genomeWhole-genome sequencing includes DNA shearing and repair before adapter ligation.Low depth or genome skimming of whole genomes is performed for organelle (plastome,mitochondrial, or rDNA), phylogenetic/systematics, or comparative analysis. Can provide partialsequences of low-copy nuclear loci for designing PCR primers or probes for subsequenthybridization-based genome reduction approaches.13OS-SeqHybridization basedOligonucleotide-selective sequencing is a targeted genome resequencing in which the lawnof oligonucleotide primers of an Illumina flow cell is modified to function as both a capture andsequence substrate.28Array hybridizationcapture (with orwithout C0t1)Hybridization basedFragment library hybridized to immobilized probe. Non-specific hybrids are removed and targetedDNA is eluted and sequenced. Can be less labor intensive than PCR amplification. Can befollowed by a target-specific array that enriches for target in a reduced-complexity sample.15,16In-solutionhybridization capture(with or without C0t1)Hybridization basedSpecific probes designed to target regions of interest from sequencing library. An excess ofprobes over template can result in a higher hybridization than with array-based methods. Canbe more amenable to scalable throughput.17Restriction digestComplexity reduction using AFLP with next-generation sequencing. Enables SNP discoveryusing tagged libraries of 2 or more genetically diverse samples. Uses a methylation-sensitiverestriction enzyme sequenced at 5–10 redundancy. Use of homozygous lines is encouragedto enable selection of SNPs located in low- or single-copy genome sequences.22RAD-SeqRestriction digestGenomic DNA digested with a restriction enzyme and a barcoded adapter is ligated tocompatible sticky ends. DNA samples, each with a different barcode, are pooled, randomlysheared, and size selected (300–700 bp), and a second adapter is ligated after polishing andfilling ends. A Y-adapter ensures that only RAD tags are amplified in the PCR step.5Cornell GBSRestriction digestEmploys unmodified adapters (ie, without the 5’ phosphate group and fork), removes fragmentsize selection. By using a single well for genomic DNA digestion and adapter ligation, it hasreduced a number of enzymatic and purification steps. Methylation-sensitive enzymes are usedto avoid repetitive regions of plant genomes.9Modified Cornell GBSRestriction digestModifies the original Cornell GBS method by use of 2 complementary enzymes (a “rare” cutterand a “common” cutter) and a Y adapter where Adapter 1 and Adapter 2 are on opposite endsof each fragment.8Restriction digestRelies on the concept of RAD-Seq, but eliminates the random shearing. Explicitly uses sizeselection to recover a tunable number of regions distributed randomly through the genome.Provides an index, computational analysis tool kit, and lightweight data management tools tofacilitate multiplexing of many hundreds of individuals. Major cost reductions are attributed toremoval of random shearing and subsequent end repair requirements.20Restriction digestGenome reduction based on restriction site conservation. Includes a double digest of DNA withrare and frequent restriction enzymes, labeling a recognition rare cutter site with 5’ biotin usingparamagnetic bead separation, adding barcode sequences using PCR, equimolar pooling ofsamples, and size selection using gel isolation.29–30MSGRestriction digestMultiplex NGS protocol, includes a fragment size-selection step developed to identifyrecombinant breakpoint of many samples simultaneously at resolution sufficient for mostmapping purposes. Incorporates aspects of WGS and RAD-Seq. Uses a more frequent cutterthan RAD-Seq and allows ligation of adapters to many small genomic fragments in a singlestep. Fragment orientation is random regarding the direction of sequencing. No shearing orrepair of DNA before adapter ligation.19DArTSeqRestriction digestBased on genome complexity reduction using restriction enzymes followed by sequencing.31Amplicon sequencingLR-PCRMolecular SC
Application Spotlight: AgrigenomicsSummaryGenotyping arrays forged the foundation of thegenomics movement in agriculture, identifyingSNPs associated with desired phenotypic traits thatresearchers have used to improve livestock breedingand crop yields. The rapid evolution of sequencingtechnologies is driving the development of lower-costsequencing-based genotyping methods that will enableagrigenomics researchers to study livestock, crops,and biological systems at a level never before possible.Providing a genome-wide view, NGG methods offerthe specificity, reproducibility, and efficiency neededto accelerate agricultural research, advance thedevelopment of high-value trait screening methods,and enable the swift deployment of these applicationsin the real world.References1. Batley, J. and D. Edwards (2007). SNP applications in plants. InOraguzie NC, Rikkerink EHA, Gardiner SE, and Silva HN (Eds.),Association Mapping in Plants, 95–102. New York, New York,USA: Springer.2. Wang DG, Fan JB, Siao CJ, et al. Large-scale identification,mapping, and genotyping of single-nucleotide polymorphisms inthe human genome. Science. 1998;280:1077-1082.12. Lorenc MT, Hayashi S, Stiller J, et al. Discovery of single nucleotidepolymorphisms in complex genomes using SGSautoSNP. Biology.2012;1:370-382.13. Huang X, Feng Q, Quian Q, et al. High-throughput genotyping bywhole-genome resequencing. Genome Res. 2009;19:1068-1076.14. Grover CE, Salmon A, Wendel JF. Targeted sequence capture asa powerful tool for evolutionary analysis. Am J Bot. 2012;99:312319.15. Fu Y, Springer NM, Gerhardt DJ, et al. Repeat subtractionmediated sequence capture from a complex genome. Plant J.2010;62:898-909.16. Mamanova L, Coffey AJ, Scott CE, et al. Target-enrichmentstrategies for next-generation sequencing. Nat Methods.2010;7:111-118.17. Saintenac C, Jiang D, Akhunov ED. Targeted analysis of nucleotideand copy number variation by exon capture in allotetraploid wheatgenome. Genome Biol. 2011;12:R88.18. Liu S, Yeh CT, Tang HM, Nettleton D, Schnable PS. Genemapping via bulked segregant RNA-Seq (BSR-Seq). PLoS One.2012;7:e36406.19. Andolfatto P, Davison D, Erezyilmaz D, et al. Multiplexed shotgungenotyping for rapid and efficient genetic mapping. Genome Res.2011;21: 610-617.20. Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE.Double digest RADSeq: an inexpensive method for de novo SNPdiscovery and genotyping in model and non-model species. PLoSOne. 2012;7:e37135.21. Nielsen R, Paul JS, Albrechtsen A, Song YS. Genotype and SNPcalling from next-generation sequencing data. Nat Rev Genet.2011;12:443-451.3. Van Tassell CP, Smith TPL, Matukumalli LK, et al. SNP discoveryand allele frequency estimation by deep sequencing of reducedrepresentation libraries. Nat Methods. 2008;5:247-252.22. Van Orsouw NJ, Hogers RC, Janssen A, et al. Complexityreduction of polymorphic sequences (CRoPS): a novel approachfor large-scale polymorphism discovery in complex genomes.PLoS One. 2007;2:e1172.4. Boichard D, Chung H, Dassonneville R, et al. Design of alow-density SNP array optimized for imputation. PLoS One.2012;7:e34130.23. Li Y, Sidore C, Kang HM, Boehnke M, Abecasis GR. Lowcoverage sequencing: implications for design of complex traitassociation studies. Genome Res. 2011;21:940-951.5. Baird NA, Etter PD, Atwood TS, et al. Rapid SNP discovery andgenetic mapping using sequenced RAD markers. PLoS One.2008;3:e3376.24. 1000 Bull Genomes Project (www.1000bullgenomes.com)Accessed 18 December 2014.6. Kirst M, Resende M, Munoz P and Neves L. Capturing andgenotyping the genome-wide genetic diversity of trees forassociation mapping and genomic selection. BMC Proceedings.2011;5:17.7. Metzger ML. Sequencing technologies—the next generation. NatRev Genet. 2010;11:31-46.8. Poland JA, Brown PJ, Sorrells ME, Jannink J. Development ofhigh-density genetic maps for barley and wheat using a noveltwo-enzyme genotying-by-sequencing approach. PLoS One.2012;7:e32253.9. Elshire RJ, Glaubitz JC, Sun Q, et al. A robust, simple genotypingby-sequencing (GBS) approach for high diversity species. PLoSOne. 2011;6:e19379.10. Cronn R, Knaus BJ, Liston A, et al. Targeted enrichment strategiesfor next generation plant biology. Am J Bot. 2012;99:291-311.11. McCouch SR, McNally KL, Wang W, Sackville Hamilton R.Genomics of gene banks: A case study in rice. Am J Bot.2012;99:407-423.25. Hardenbol P, Baner J, Jain M, et al. Multiplexed genotyping withsequence-tagged molecular inversion probes. Nat Biotechnol.2003;21:673-678.26. Hardenbol P, Yu F, Belmont J, et al. Highly multiplexed molecularinversion probe genotyping: over 10,0
including Illumina TruSeq Custom Amplicon. Hybridization-Based Methods Hybridization-based approaches include solid substrate as well as liquid hybridization methods, using oligonucleotide specificity to bind to and isolate complementary sequences. To leverage sequencing capacity and optimize costs, these methods rely on multiplexing samples enriched using the same probe sets. Solid phase .