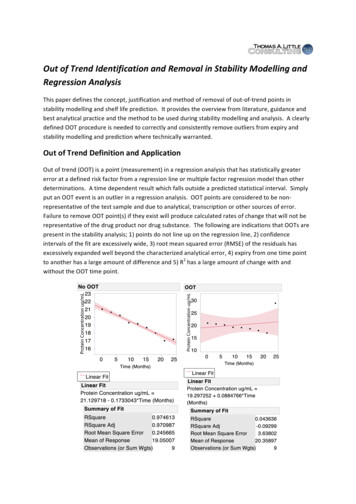
Transcription
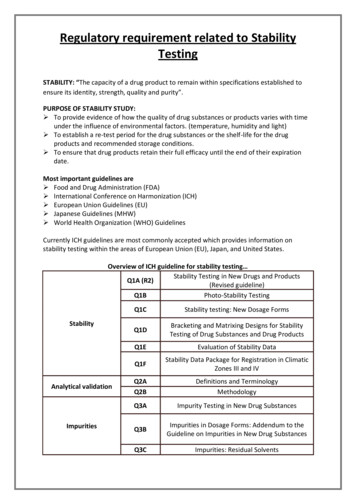
Regulatory requirement related to StabilityTestingSTABILITY: “The capacity of a drug product to remain within specifications established toensure its identity, strength, quality and purity”.PURPOSE OF STABILITY STUDY: To provide evidence of how the quality of drug substances or products varies with timeunder the influence of environmental factors. (temperature, humidity and light) To establish a re-test period for the drug substances or the shelf-life for the drugproducts and recommended storage conditions. To ensure that drug products retain their full efficacy until the end of their expirationdate.Most important guidelines are Food and Drug Administration (FDA) International Conference on Harmonization (ICH) European Union Guidelines (EU) Japanese Guidelines (MHW) World Health Organization (WHO) GuidelinesCurrently ICH guidelines are most commonly accepted which provides information onstability testing within the areas of European Union (EU), Japan, and United States.Overview of ICH guideline for stability testing Stability Testing in New Drugs and ProductsQ1A (R2)(Revised guideline)Q1BPhoto-Stability TestingStabilityAnalytical validationImpuritiesQ1CStability testing: New Dosage FormsQ1DBracketing and Matrixing Designs for StabilityTesting of Drug Substances and Drug ProductsQ1EEvaluation of Stability DataQ1FStability Data Package for Registration in ClimaticZones III and IVQ2AQ2BDefinitions and TerminologyMethodologyQ3AImpurity Testing in New Drug SubstancesQ3BImpurities in Dosage Forms: Addendum to theGuideline on Impurities in New Drug SubstancesQ3CImpurities: Residual Solvents
PharmacopeiasQ4Pharmacopeial harmonizationBiotechnologyQualityQ5AViral Safety EvaluationQ5BGenetic StabilityQ5CStability of Biotechnology ProductsQ5DCell SubstratesQ6ASpecifications, Test Procedures, andAcceptance Criteria for New DrugSubstances and ProductsQ6BBiotechnological substanceGMPQ7AGMP for active pharmaceutical ingredientsDevelopmentQ8Pharmaceutical developmentManagementQ9Quality Risk ManagementSpecificationSTABILITY E1EFFICACY GUIDELINESE2(A)E2(B)E2CGuidelines on The Need For Carcinogenicitystudies of pharmaceuticalsTesting for carcinogenicity of pharmaceuticalsDose selection for carcinogecity studies ofpharmaceuticalsGuidance on specific aspect of regulatorygenotoxicity test for pharmaceuticalsStandard for genotoxicity testing forpharmaceuticalsNote for guidance on ToxicokineticsPharmacokinetic:- Guidance for repeated dosetissue distribution studiesDuration of Chronic Toxicity testing in AnimalsDetection of Toxicity To Reproduction forMedicinal product and toxicity to male fertilityPreclinical Safety Evaluation Of Biotechnologyderived PharmaceuticalsSafety Pharmacology studies for HumanPharmaceuticalsImmunotoxicity studies for HumanPharmaceuticalsThe Extent of Population Exposure to Assessclinical SafetyClinical safety Data managementImplementation working groupClinical safety Data management :- periodic safety
IPLINEGUIDELINESM1M2M3update reports & marketed drugsPost aproval safety data manegemant :Definations and Standards for expedited reportingPharmacovigillance PlanningDevelopment safety update reportStructure and content of clinical study reportsDose response Information to support drugregisitrationEthnic factors in the acceptability of foreignclinical dataGuideline for Good Clinical PracticeStudies in support of Specific PopulationGeneral Consideration For Clinical TrialsStastical Principles For Clinical TrialsClinical Investigation of medicinal products In ThePediatric populationPrinciples Of Clinical Evaluation of New Antihypertensive drugsThe Clinical Evaluation of proarrythmic potentialfor Non-Antiarrythmic drugsDefinations of genomic biomarkers,pharmacoecononomics, pharmacogenetics,Genomic DATA & sample coding categoriesMaintenance of The ICH Guideline on non-clinicalsafety studies for the conduct of human clinicaltrials for pharmaceuticalsElectronic Transmission of Individual Case SafetyReports Message SpecificationOrganisation of the Common Technical Documentfor the Registration of Pharmaceuticals for HumanUseINTERNATIONAL CLIMATIC ZONES AND CLIMATIC CONDITIONSClimaticConditionZone ITemperateZone IIMediterranean(sub-tropical)Zone IIIZone IVHot/dry orVeryHot/moderate hot/humidRHMean Annual 20 CTemperature20.5-24 C 24 C 24 CKinetic Mean 21 CTemperature(Virtualtemperature)26 C31 C31 C
Mean Annual 45%RelativeHumidity60%40%70%REQUIREMENT OF TEMPERATURE DEPENDED ON TYPE OF TESTINGTYPE OF STUDYLong termIntermediateAcceleratedTEMPERATURE25 C 2 C30 C 2 C40 C 2 C/RELATIVEHUMIDITY/60% RH 5% RH/65% RH 5% RH75% RH 5% RHTIME DURATION12 months6 months6 monthsDIFFERENT TEMPERATURE REQUIREMENT DEPEND UPON TYPE OF DOSAGEFORMSFOR DISTINCTPRODUCTSSolid oral DF, solids forreconstitution, dry&lyophilized powders inglass vialsLiquids in glass bottles,vials, sealed glassampoules which provide animpermeable barrier towater lossDrug products insemipermeable containersTYPE OF STUDYAST40 C 2 C75 % 5%RH40 C 2 CAmbientHumidity40 C 2 CNMT 25 %RHIST40 C 2 C75 % 5% RH30 C 2 CAmbienthumidity30 C 2 C65 % 5%RHLST40 C 2 C75 % 5% RH25 C 2 CAmbientHumidity25 C 2 C40 % 5% RHOr 30 C 2 C35 % 5% RHSUPAC GUIDELINES1) Stability Testing for New Drug Applications(NDA)A. Drug SubstanceB. Drug Product2) Stability Testing for Abbreviated New Drug Applications(ANDA)A. Drug Substance Stability Data Submission Supporting information may be provided directly to the drug product ANDA or byreference to an appropriately referenced drug master file (DMF). For ANDA bulk drug substances- on a minimum of one pilot-scale batch. ANDA bulk drug substances produced by fermentation- on three production batches,at least two of which should be generated from different starter cultures.B. Drug Substance Testing
A program for stability assessment may include storage at accelerated, long-term,and, if applicable, intermediate stability study storage conditions (refer to IV.G. ofthe ICH Q1A Guidance and Section II.A. of this guidance).C. Drug Product As per ICH Q1 A [Section II.B.]D. ANDA Data Package Recommendations Accelerated stability data at 0, 1, 2, and 3 months. A tentative expiration datingperiod of upto 24 months will be granted based on satisfactory accelerated stabilitydata unless not supported by the available long-term stability data. Long-term stability data Additional stability studies accelerated stability study.E. Stability Study Acceptance3) Stability Testing For Investigational New Drug Applications The amount of information needed to achieve that assurance will vary witho The phase of the investigation,o The proposed duration of the investigation,o The dosage form.A. General Supportive stability data for changes to an approved drug application (i.e. postapproval changes) required . If change does not alter the stability of the drug product, the previously approvedexpiration dating period can be used. But now SUPAC-IR, MR , SS guidance are followed for stability studies . Provides 5 stability data package types .B. Change in Manufacturing Process of the Drug Substance Carried out at approved manufacturing site . Should be supported by the submission of sufficient data to show that such changedoes not compromise the quality , purity , or stability of the drug substance and theresulting drug product Special concerns are there for biological products.C. Change in Manufacturing SiteSite changes consists of change in location site of : Manufacture Packaging operations Analytical testing laboratory both of company owned and contractmanufacturing. Sufficient data to show that such a change does not alter the characteristics orcompromise the quality, purity, or stability of the drug substance or drug productmay be necessary. The data should include a side-by-side comparison of all attributes to demonstratecomparability and equivalency of the drug substance or drug product manufacturedat the two facilities. New manufacturing locations should have a satisfactory cGMP inspection.D. Change in Formulation of the Drug Product Historically, all changes in drug product formulation were grouped together andrequired extensive stability documentation, usually submitted as a prior-approvalsupplement.
An exception was the detection of a color from a product that could be reported inan annual report without supporting stability dataE. Addition of a New Strength for the Drug Product The addition of a new strength for an approved drug product will generallyrequire the submission of a prior-approval supplement. Demonstration of equivalent stability between the approved drug product and thenew strength will allow extension of the approved drug product expiration dating tothe new strength. New strengths intermediate to those of an approved drug product may be supportedby bracketing/Matrixing studies (See Section VII.G. and VII.H.).F. Change in Manufacturing Process and/or Equipment for the Drug Product Can be supported by the submission of sufficient data to show that such a changedoes not alter the characteristics or compromise the stability of the drug product. The standard stability commitment to conduct and/or complete the stability studieson the first three production batches produced by the revised manufacturingprocess in accordance with the approved stability protocol is necessary. If the data are found acceptable, the approved expiration dating period may beretained.G. Change in Batch Size of the Drug Product A key question : whether the change involves a change in equipment or its mode ofoperation, or other manufacturing parameters described for the approved batchsize. Table 19 presents the recommended stability data packages for a variety of batchsize situations not involving equipment or mode of operation changes. If an equipment change is part of the batch size change, please refer to Change inManufacturing Process of the Drug Product (Section IX.F.).H. Reprocessing of a Drug Product Stability data submitted should take into account the nature of the reprocessingprocedure and any specific impact that might have upon the existing stability profileof the drug. The expiration dating period for a reprocessed batch should not exceed that of theparent batch, and the expiration date should be calculated from the original date ofmanufacture of the oldest batch. Reprocessing range from repackaging to regrinding and recompressing tablets. Any batch of the drug product that is reprocessed should be placed on acceleratedand long-term stability studies using the approved protocol to generate a Type 2stability data package.I. Change in Container and Closure of the Drug Product The first factor used in determining the stability data package recommendation iswhether or not the protective properties of the container/closure system areaffected by the proposed change. Protective properties of the container/closure system include,Moisture permeability,Oxygen permeability,Light transmission. Changes that may affect these properties should be supported by a greater amountof data to support the change. The second factor is the nature of the dosage form itself. A solid dosage form willgenerally be less affected by a container change than a liquid dosage form
PHOTOSTABILITY TESTING OF NEW DRUG SUBSTANCES ANDPRODUCTS1.GENERAL The ICH Harmonized Tripartite Guideline covering the Stability Testing of New DrugSubstances and Products notes that light testing should be an integral part of stresstesting.A. Preamble The intrinsic photostability characteristics of new drug substances and products shouldbe evaluated to demonstrate that, as appropriate, light exposure does not result inunacceptable change. Normally, photostability testing is carried out on a single batch of material. Under some circumstances these studies should be repeated if certain variations andchanges are made to the product (e.g., formulation, packaging). The guideline primarily addresses the generation of photostability information forsubmission in Registration Applications for new molecular entities and associated drugproducts. The guideline does not cover the photostability of drugs after administration(i.e. under conditions of use). A systematic approach to photostability testing is recommended covering, asappropriate, studies such as:i) Tests on the drug substance;ii) Tests on the exposed drug product outside of the immediate pack;And if necessary;iii) Tests on the drug product in the immediate pack;And if necessary;iv) Tests on the drug product in the marketing pack. The formal labeling requirements for photo labile drug substances and drug productsare established by national/regional requirements.B. Light Sources The light sources described below may be used for photo stability testing. The applicant should maintain an appropriate control of temperature to minimize theeffect of localized temperature changes.Option 1 Any light source that is designed to produce an output similar to the D65/ID65 emissionstandard such as an artificial daylight fluorescent lamp combining visible and ultraviolet(UV) outputs, xenon, or metal halide lamp. D65 is the internationally recognized standard for outdoor daylight as defined in ISO10977 (1993). ID65 is the equivalent indoor indirect daylight standard. For a light source emitting significant radiation below 320 nm, an appropriate filter(s)may be fitted to eliminate such radiation.Option 2 For option 2 the same sample should be exposed to both the cool white fluorescent andnear ultraviolet lamp.
1. A cool white fluorescent lamp designed to produce an output similar to thatspecified in ISO 10977(1993) ; and2. A near UV fluorescent lamp having a spectral distribution from 320 nm to 400 nmwith a maximum energy emission between 350 nm and 370 nm; a significantproportion of UV should be in both bands of 320 to 360 nm and 360 to 400 nm.C. Procedure For confirmatory studies, samples should be exposed to light providing an overallillumination of not less than 1.2 million lux hours and an integrated near ultravioletenergy of not less than 200 watt hours/square meter to allow direct comparisons to bemade between the drug substance and drug product.DECISION FLOW CHART FOR PHOTOSTABILITY TESTING OF DRUG PRODUCTS2. DRUG SUBSTANCE For drug substances, photostability testing should consist of two parts: forceddegradation testing and confirmatory testing. The purpose of forced degradation testing studies is to evaluate the overallphotosensitivity of the material for method development purposes and/or degradationpathway elucidation. This testing may involve the drug substance alone and/or in simplesolutions/suspensions to validate the analytical procedures. In these studies, the samples should be in chemically inert and transparent containers.
In these forced degradation studies, a variety of exposure conditions may be used,depending on the photosensitivity of the drug substance involved and the intensity ofthe light sources used. For development and validation purposes it is appropriate to limit exposure and end thestudies if extensive decomposition occurs. For photo stable materials, studies may be terminated after an appropriate exposurelevel has been used. Under forcing conditions, decomposition products may be observed that are unlikely tobe formed under the conditions used for confirmatory studies. Confirmatory studies should then be undertaken to provide the information necessaryfor handling, packaging, and labeling. Normally, only one batch of drug substance is tested during the development phase, andthen the photo stability characteristics should be confirmed on a single batch if the drugis clearly photo stable or photo labile. If the results of the confirmatory study are equivocal, testing of up to two additionalbatches should be conducted. Samples should be selected as described in the Parent Guideline.A. Presentation of Samples Care should be taken to ensure that the physical characteristics of the samples undertest are taken into account and efforts should be made, such as cooling and/or placingthe samples in sealed containers, to ensure that the effects of the changes in physicalstates such as sublimation, evaporation or melting are minimized. All such precautions should be chosen to provide minimal interference with theexposure of samples under test. Possible interactions between the samples and anymaterial used for containers or for general protection of the sample, should also beconsidered and eliminated wherever not relevant to the test being carried out. As a direct challenge for samples of solid drug substances, an appropriate amount ofsample should be taken and placed in a suitable glass or plastic dish and protected witha suitable transparent cover if considered necessary. Solid drug substances should be spread across the container to give a thickness oftypically not more than 3 millimeters. Drug substances that are liquids should be exposed in chemically inert and transparentcontainers.B. Analysis of Samples At the end of the exposure period, the samples should be examined for any changes inphysical properties (e.g., appearance, clarity, or color of solution) and for assay anddegradants by a method suitably validated for products likely to arise fromphotochemical degradation processes. Where solid drug substance samples are involved, sampling should ensure that arepresentative portion is used in individual tests. Similar sampling considerations, suchas homogenization of the entire sample, apply to other materials that may not behomogeneous after exposure. The analysis of the exposed sample should be performed concomitantly with that of anyprotected samples used as dark controls if these are used in the test.C. Judgement of Results The forced degradation studies should be designed to provide suitable information todevelop and validate test methods for the confirmatory studies.
These test methods should be capable of resolving and detecting photolytic degradantsthat appear during the confirmatory studies. When evaluating the results of these studies, it is important to recognize that they formpart of the stress testing and are not therefore designed to establish qualitative orquantitative limits for change. The confirmatory studies should identify precautionary measures needed inmanufacturing or in formulation of the drug product, and if light resistant packaging isneeded. When evaluating the results of confirmatory studies to determine whether change dueto exposure to light is acceptable, it is important to consider the results from otherformal stability studies in order to assure that the drug will be within justified limits attime of use (see the relevant ICH Stability and Impurity Guidelines).3. DRUG PRODUCT(It is same as that described in drug substances)4.ANNEXA. Quinine Chemical Actinometry The following provides details of an actinometric procedure for monitoring exposure toa near UV fluorescent lamp (based on FDA/National Institute of Standards andTechnology study). For other light sources/actinometric systems, the same approach may be used, but eachactinometric system should be calibrated for the light source used. Prepare a sufficient quantity of a 2 per cent weight/volume aqueous solution of quininemonohydrochloride dihydrate (if necessary, dissolve by heating).Option 1: Use 20 ml colourless ampoules (seal hermetically).Shape and Dimensions for ampoule specifications.Option 2: Use 1 cm quartz cell. For both the options, prepare sample and control wrap in aluminum foil to protectcompletely from light, and measure their absorbance At and Ao respectively at 400nmusing 1cm path length. Measure the change in absorbance. The length of exposure should be sufficient to ensure a change in absorbance of at least0.9.
BRAC
A program for stability assessment may include storage at accelerated, long-term, and, if applicable, intermediate stability study storage conditions (refer to IV.G. of the ICH Q1A Guidance and Section II.A. of this guidance). C. Drug Product As per ICH Q1