Transcription
CTD Dossier PreparationK. Srikantha ReddySr.Manager-Regulatory AffairsMedreich LimitedSrikanth.k@medreich.com
CTD Dossier Preparation CTD (Common Technical Document)contains 5 modules Module – 1Module – 2Module – 3Module – 4Module – 5
DMFDrug Master File (DMF) is a submission to theFood and Drug Administration (FDA) that maybe used to provide confidential detailedinformation about facilities, processes, orarticles used in the manufacturing, processing,packaging and storing of one or more humanpackaging,drugs. The information contained in the DMFmay be used to support following,––––Investigational New Drug Application (IND),(IND)New Drug Application (NDA),Abbreviated New Drug Application (ANDA),Export Application.
ANDA: An Abbreviated New Drug Application (ANDA)is an application for a U.S. generic drugapprovall forf an existingi tililicensedd medicationdi tiorapproved drug. The ANDA contains data which when submittedto FDA's Center For drug Evaluation andResearch (CDER), Office of Generic Drugs,provides for the review and ultimate approvalpppof a generic drug product. Once approved, anapplicant may manufacture and market thegeneric drug product to provide a safe,effective, low cost alternative to the Americanpublic.
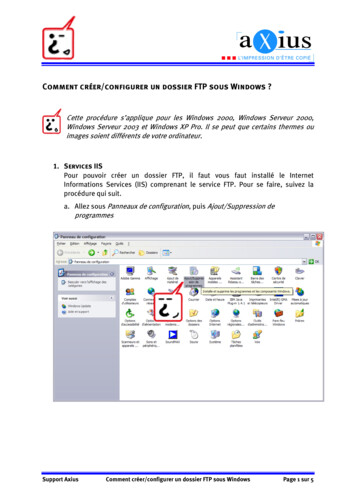
Module – 1(e.g. g Information1 0 Cover Letter1.01.1 Comprehensive Table of Content1.2 Application Form1 3 Product1.3P d t IInformationfti1.3.1 SPC’s, Labelling and Packaging1.3.2 Mock-Up1.3.3 Specimen1.3.4 Consultation with target patient group1 3 5 SPC1.3.5SPC’ss already approved in the Memberstates1.3.6 Braille
Module - 11.4Information about the Experts1.5SpecificpRequirementsqtypes of applications1.61.7Environmental Risk Assessmentg to OrphanpMarketInformation gilanceInformation relating to Clinical Trials1.81.9fordifferent1.10 Information relating to Pediatrics1.11 Response to Queries1.12 Additional Data
Module - 2Module - 2: CTD Summary2.1212.22.32.42.52.62.7TableTbl off CContentt t (Comprehensive)(Chi al, including its pharmacology class,moded off action,tiandd proposedd clinicalli i l use))Quality Overall SummaryNon-clinical OverviewClinical OverviewNon-clinical Written and Tabulated SummariesyClinical summary
Module - 22.4 Non-clinical OverviewGenerale a Aspectsspects2.4.1 Ge2.4.2 Content and Structural Format2.5 Clinical Overview2.5.1 Product Development of Content Rationale2.5.2 Overview of Biopharmaceutics2 5 3 Overview of Clinical Pharmacology2.5.32.5.4 Overview of Efficacy2.5.5 Overview of Safety2.5.6 Benefits and Risks Conclusions2.5.7 Literature References
Module - 22.6 Non-clinical Written and Tabulated Summaries2.6.1 Pharmacology2 6 2 Pharmacokinetics2.6.2Phki ti2.6.3 Toxicology2 7 Clinical summary2.72.7.1 Biopharmaceutic Studies and AssociatedAnalytical Methods2.7.2 Clinical Pharmacology Studies2.7.3 Clinical Efficacy2 7 4 Clinical2.7.4Cli i l SSafetyf t2.7.5 Literature References2 7 6 Synopses of Individual Studies2.7.6
Module - 3Module – 3: QualityQy3.1 Table of Contents3.2 Bodyy of Data3.2.S Drug Substance3.2.S.1 General Information3.2.S.1.1 Nomenclature3.2.S.1.2 Structure3 2 S 1 3 General Properties3.2.S.1.3
Module - 33.2.S.2 Manufacture3.2.S.2.1 Manufacturer Details3.2.S.2.2 Description of Manufacturing Process andProcess Controls3 2 S 2 3 Control of Materials3.2.S.2.33.2.S.2.4 Controls of Critical Steps andIntermediates3 2 S 2 5 Process3.2.S.2.5PValidationV lid ti andd /or/ EvaluationE l ti3.2.S.2.6 Manufacturing Process Development3.2.S.3 Characterisation3.2.S.3.1 Elucidation of structure and otherCharacteristics3.2.S.3.233 Impuritiespu
Module - 33.2.S.4 Control of Drug Substance3.2.S.4.1 Specification of Drug Substance3.2.S.4.2 Analytical Procedures3.2.S.4.3 Validation of Analytical Procedures3.2.S.4.4 Batch Analyses3.2.S.4.5 Justification of Specification3 2 S 5 Reference Standards or Materials3.2.S.53.2.S.6 Container Closure System3.2.S.7 Stability3.2.S.7.1 Stability Summary and Conclusions3.2.S.7.2 Post-approval Stability Protocol andStability Commitment3.2.S.7.3 Stability Data
Module - 33.2.P Drug Product3.2.P.1 Description and Composition of the DrugProduct3 2 P 2 Pharmaceutical3.2.P.2Phti l DDevelopmentlt3.2.P.2.1 Components of Drug Product3 2 P 2 2 Drug Product3.2.P.2.23.2.P.2.3 Manufacturing ProcessDevelopmentp3.2.P.2.4 Container Closure System3.2.P.2.5 Microbiological Attributes3.2.P.2.6 Compatibility
Module - 33 2 P 3 Manufacture3.2.P.3M f t3.2.P.3.1 Manufacturer3 2 P 3 2 Batch Formula3.2.P.3.23.2.P.3.3 Description of Manufacturing Processand Process Controls3.2.P.3.4 Controls of Critical Steps andIntermediates3 2 P 3 5 Process Validation and /or Evaluation3.2.P.3.5
Module - 33 2 P 4 Control of Excipients3.2.P.43.2.P.4.1 Specifications3 2 P 4 2 Analytical Procedures3.2.P.4.23.2.P.4.3 Validation of AnalyticalProcedures3.2.P.4.4 Justification of Specifications3 2 P 4 5 Excipients of Human or Animal3.2.P.4.5Origin3.2.P.4.6 Novel Excipients
Module - 332P5 C3.2.P.5Controlt l off DDrug ProductP d t3.2.P.5.1 Specification of Drug Product3 2 P 5 2 Analytical Procedures3.2.P.5.23.2.P.5.3 Validation of Analytical Procedures3.2.P.5.4 Batch Analyses3.2.P.5.5 Characterisation of Impuritiesp3.2.P.5.6 Justification of Specification3.2.P.6 Reference Standards or Materials3.2.P.7 Container Closure System
Module - 33.2.P.8 Stability3.2.P.8.1 Stability Summary andC l iConclusions3.2.P.8.2 Post-approvalStabilityProtocol and StabilityCommitment3 2 P 8 3 Stability Data3.2.P.8.3
Module - 33.2.A Appendices3.2.A.1 Facilities and Equipment3.2.A.2 Adventitious Agents Safety Evaluation3 2 A 3 Novel Excipients3.2.A.33.2.R Regional Information/ Requirements3.2.R.1 Process Validation and or Evaluation3.2.R.2 Medical Device3.2.R.3 Restricted part of DMF3 2 R 4 Medicinal3.2.R.4M di i l productsd t containingt i ior usingiiinthe manufacturing process materials ofanimal and / or human origin.3.3 List of Literature References
Module - 4Module - 4: Non-clinical Studyy Reportsp4.1Table of contents4.2Study Reports4 2 1 Pharmacology4.2.14.2.1 Primary Pharmacodynamic4.2.2 Secondary Pharmacodynamic4 2 3 Safety4.2.3S f t pharmacologyhl4.2.4 Pharmacodynamic druginteractions
Module - 44.2.2 Pharmacokinetics4.2.2.1 Analytical Methods andvalidation Reports4 2 2 2 Absorption4.2.2.24.2.2.3 Distribution4.2.2.4 Metabolism4.2.2.5 Excretion4.2.2.6 Pharmacokinetic DrugInteractions4.2.2.7 Other Pharmacokinetic studies
Module - 44.2.3 Toxicology4 2 3 1 Single-dose toxicity4.2.3.14.2.3.2 Repeat-dose toxicity4.2.3.3 Genotoxicityy4.2.3.4 Carcinogenicity4.2.3.5 Reproductive and developmentalt i ittoxicity4.2.3.6 Local tolerance4 2 3 7 Other toxicity studies4.2.3.74.3 Literature References
Module - 5M d l - 5:Module5 ClinicalCli i l StudySt d ReportsRt5.15.25.3Table of ContentsTabular Listings of All Clinical StudiesClinical Study Reports5.3.1.1 Bioavailability (BA) study Reports5.3.1.2 Comparative BA andBioequivalence study reports5.3.1.3 In-vitro In-vivo Correlation studyreports5.3.1.4 Reports of Bioanalytical andAnalytical methods5 3 2 1 Plasma Protein Binding Study5.3.2.1Reports5.3.2.2 Reports of Hepatic metabolismand Drug Interaction Studies5.3.2.3 Reports of Studies Using humanBiomaterials
Module - 55331 H5.3.3.1Healthylth SSubjectbj t PK andd InitialI iti lTolerability study reports5.3.3.2 Patient PK and Initial Tolerabilityystudy reports.5.3.3.3 Intrinsic Factor PK study reports5 3 3 4 Extrinsic5.3.3.4E t i i FFactort PK studyt d reportst5.3.3.5 Population PK study reports5 3 4 1 Healthy subject PD and PK/PD5.3.4.1study reports5.3.4.2 Patient PD and PK/PD studyreports5.3.5.1 Study reports of controlled clinicalstudies
Module - 55.3.5.2 Study reports of Uncontrolled clinicalstudies5.3.5.3 Reports of Analyses of data frommore than one study5.3.5.4 Other clinical study reports5.3.6Reports of Post-MarketingEExperiencei5.3.7Case report forms and Individualpatient listings5.4 List of Key Literature References
eCTD(Version 3.2.2)06.05.2011K. Srikantha ReddySr. ManagerManager-Regulatory AffairsMedreich LimitedSrikanth.k@medreich.com
eCTD eCTD – electronic Common Technical Document The eCTD is the electronic equivalent to the CTD.CTD Regulatory Perspective “The eCTD is defined as an interface for industry toagency transfer of regulatory information while atthe same time takingg into consideration thefacilitation of the creation, review, lifecyclemanagement and archival of the electronicsubmission ”submission. Common structure for Modules 2 through 5 Agency specific requirements for Modules 1
eCTD Technical Perspective Structured set of common folders structurecontaining PDFs and SAS files (Statistical AnalysisSoftware) on a CD/DVD (Can also be submittedthrough Agency web portals) The eCTD backbone is an XML file (ExtensibleM k LMarkupLanguage)) representingti theth structuret toff thethsubmission, it includes links to files and othermetadata such as check sum information. Theschema for the XML is very rigid. PDF hyperlinks
eCTD Granularity of files submitted is small (there are no longerissues of creating large volumes of PDFs). Increased potential for reusing the same submissioncontent across agency submissions. The standard, and many of the modules have been agreedupon by the main worldwide agencies. Once a submission is sent in eCTD format all futuresubmissions for the application should be in eCTD format. OpportunityOt it tot use PartP t 11 CompliantCli t ElectronicEl t iSignatures. Use only file formats specified in the guidance
eCTD Benefits Easy to distribute and review More efficient use of resources, less cost and stress tothe organization Highly organized electronic table of contents Searchable Self-validating Integrated document and life-cycle management Cross submission integration Living document New, replace, append & delete
How it is different to Paper/Document CTD Overall Table of contents provided in XML (ExtensibleMarkup Language) Utility files to enable technical conformance andviewingi i Submission Folders, XML and Utility Files are createdautomaticallytti ll if an eCTDCTD builderb ild isi used.d Generally high level of granularity in documents Structure is more precise Lifecycle Management of the submission is easier.
eCTD Implementation - FDA Jan 1, 2008, eCTD became CDER’s standard forelectronic submission. FDA has made it mandatory for all ELECTRONICsubmissionsb i ito beb ini eCTD formatfsincei2007-08.However, paper copies are still accepted. Suitablewaivers will have to be taken before hand. The number of ANDA submissions to FDA hasincreased from 72 in the year 2006 to 1550 in 2009
eCTD Implementation - EU( p(http://esubmission.emea.europa.eu/)p) Requirements on Electronic submissions (Nees (Non-eCTD electronicsubmission, Version 2.0 March-2010) and eCTD) and paper documentation forNew Application within MRP, DCP or National procedure –Refer CMDh/085/2008/Rev7 October 2010) FromF11stt JulyJ l 2010,2010 ththe EU M1 v1.41 4 mustt bbe usedd ffor alllleCTD submissions for all European procedures,
eCTD Implementation - Marketingauthorishtt//hk/Phti li d t /M k tith iations/index.htm The preferred format for new marketing authorization (MA)applicationsli iiis theh electronicli CCommon TTechnicalh i l DossierD i (eCTD)( CTD) eCTD applications must be created according to the currentspecifications: eCTD specification v 3.2.2 MHRA will accept applications in PDF-only format (Note that all PDFfiles included in an eCTD (irrespective of the module) should be v1.4, exceptwhere there is an agency-specific requirement for a later version (e.g. for anapplication form)). The Summary of Product Characteristics (SmPC) will need to beprepared using the Word template. Use the MHRA Adobe Application form which is available via theMHRA Portal. This will produce an XML file that MHRA can uploaddirectly into their database.database
Regulatory Contact information
eCTD Modules When making an electronic submission, each documentshould be pprovided as a separatepfile. The documents, whether for a marketing application, aninvestigational application, or a related submission,should be organized based on the five modules in theCTD: Moduled l 1 includesi l d administratived i ii informationi fi anddprescribing information, Mod le 2 inclModuleincludesdes CTD summarys mmar documents,doc ments Module 3 includes information on quality, Module 4 includes the nonclinical study reports, and Module 5 includes the clinical study reports.
eCTD Template
eCTD Screen Shot
eCTD Screen Shot of Module 2
eCTD Screen Shot of Module 3
eCTD Screen Shot of Module 4
eCTD Screen Shot of Module 5
eCTD Screen Shot of Module 5
eCTD Screen Shot of Module 5
eCTD Management Software eCTDXPress – Image Solutions –http://wwwimagesolutions comimagesolutions.com MasterControl Submissions Gateway - MasterControl http://www.mastercontrol.comControl,http://www mastercontrol com Liquent’s EZsubs software solution,http://www.liquent.com/pq Data Farm, http://www.datafarminc.com/ Take solution : www.PharmaReady.comwww PharmaReady com Lorenz Life Sciences : www.lorenz.cc
Thank YouSRIKANTH.K
Sr.Manager-Regulatory Affairs Medreich Limited Srikanth.k@medreich.com. CTD Dossier Preparation CTD (Common Technical Document) contains 5 modules Module – 1 Module – 2 Module – 3 Module – 4 Modul