Transcription
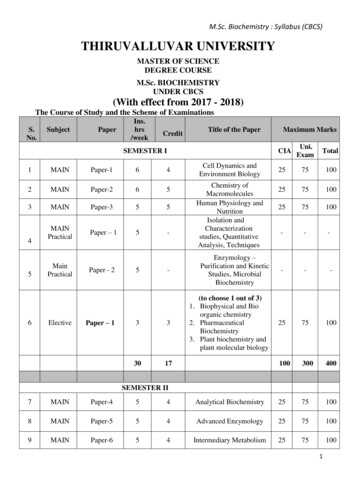
Introduction to Cell & Molecular BiologyTechniques :ImmunofluorescenceDAPI (DNA)GFPγ TubulinMerge 2014 State of Queensland (Department of Education, Training and EmploymentSPARQ-ed – University of Queensland Diamantina InstituteUniversity of Queensland, Australia.Page 1http://www.di.uq.edu.au/SPARQ-edph. 61 7 3443 6920 fax. 61 7 3443 6966Email: sparqed@uq.edu.au
SPARQed is a collaboration between The University of Queensland’s Diamantina Institute and The QueenslandGovernment’s Department of Education, Training and Employment.The Immunofluorescence Workshop is based on technique used in the SPARQ-ed Research ImmersionPrograms developed by scientists at the University of Queensland Diamantina Institute. The experimentalprotocols and all supporting materials were adapted for student use by Dr Peter Darben, under the supervisionof Dr Sandrine Roy.Cover Image : A field of HeLa cells stained with DAPI (which binds to DNA and fluoresces blue) and treated withan antibody against γ Tubulin and a secondary antibody conjugated to a red flurophore. Some of these cellshave been transfected with the gene for the jellyfish protein green fluorescent protein (GFP). The figure showsthe three colour channels, imaged separately for blue, green and red, and the combined image obtained whenthese three channels are merged. Image courtesy Rose Boutros.All materials in the manual are Copyright 2014, State of Queensland (Department of Education and Training).Permission is granted for use in schools and other educational contexts. Permission for use and reproductionshould be requested by contacting Peter Darben at p.darben@uq.edu.au. These materials may not be used forcommercial purposesRisk assessments were developed with the assistance of Paul Kristensen, Maria Somodevilla-Torres and JaneEasson.Many thanks to all in the “Gab Lab” whose patience and support made this happen. 2014 State of Queensland (Department of Education, Training and EmploymentSPARQ-ed – University of Queensland Diamantina InstituteUniversity of Queensland, Australia.Page 2http://www.di.uq.edu.au/SPARQ-edph. 61 7 3443 6920 fax. 61 7 3443 6966Email: sparqed@uq.edu.au
Table of ContentsIntroductionWhat is Cell Biology ? . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .4Common Cell Biology Techniques . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .4The WorkshopGetting Started . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .6Theoretical Basis of the ProjectTissue Culture and Cancer Cell Lines . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .6Preparation of Cells for Immunofluorescence . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .7Antibodies and Immunofluorescence . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .9Targets . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .13Fluorescence Microscopy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .14Experimental ProtocolsHow to Use this Manual . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .19Preparation of Culture Plates . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .19Blocking and Permeablisation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .19Treatment with Antibodies . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .20Imaging Using Epifluorescence Microscopy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .21What to Look Out For . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .21AppendicesAppendix A : DNA . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .22Appendix B : Proteins . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .24Appendix C : Using a Micropipette . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .29Appendix D : Glossary of Terms . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .34 2014 State of Queensland (Department of Education, Training and EmploymentSPARQ-ed – University of Queensland Diamantina InstituteUniversity of Queensland, Australia.Page 3http://www.di.uq.edu.au/SPARQ-edph. 61 7 3443 6920 fax. 61 7 3443 6966Email: sparqed@uq.edu.au
IntroductionWhat is Cell Biology ?Cells form the basis of all living things. They are the smallest single unit of life, from the simplest bacteria toblue whales and giant redwood trees. Differences in the structure of cells and the way that they carry out theirinternal mechanisms form the basis of the first major divisions of life, into the three kingdoms of Archaea(“ancient” bacteria), Eubacteria (“modern” bacteria) and Eukaryota (everything else, including us). Anunderstanding of cells is therefore vital in any understanding of life itself.Cell biology is the study of cells and how they function, from the subcellular processes which keep themfunctioning, to the way that cells interact with other cells. Whilst molecular biology concentrates largely on themolecules of life (largely the nucleic acids and proteins), cell biology concerns itself with how these moleculesare used by the cell to survive, reproduce and carry out normal cell functions.In biomedical research, cell biology is used to find out more about how cells normally work, and howdisturbances in this normal function can result in disease. An understanding of these processes can lead totherapies which work by targeting the abnormal function.Common Cell Biology TechniquesThe following list covers some of the more commonly used cell biology techniques – it is by no meansexhaustive.Cell / Tissue Culture – in the same way that bacteria and other simple organisms can be grown in thelaboratory outside their normal environment, cells and tissues from more complicated organisms canbe cultured as well. The techniques are slightly different, and the culture media are more complex toreflect the complex internal environment inside the host from which the cells are derived, howevercell and tissue culture is a powerful tool which provides an almost limitless supply of test material forresearchers to use without resorting to using whole organisms. In addition, the controlled conditionsin cell and tissue culture allow researchers to carry out experiments with a lower number of variableswhich may affect the outcome of the test. Cell culture may use cells removed directly from anorganism (primary culture), or it may use lines of cultured cancer cells. The benefit of the latterapproach is that cancer cells continue to divide, while primary cultures cease dividing after a numberof cycles.Microscopy – the basic tool of cell biology is microscopy. Recent advances in imaging technology haveallowed an unprecedented amount of information to be gleaned from microscopic analysis. Types ofmicroscopic techniques which are used include :Brightfield – traditional microscopy, where cells are illuminated by visible light. Brightfieldmicroscopy gives a general picture of cell function, although that information is not verydetailed or specific. As animal cells lack cell walls, brightfield microscopy may use specialtechniques such as phase contrast to show cellular structures in more detail. Brightfieldmicroscopy allows imaging of live or fixed (dead) cells and tissues) 2014 State of Queensland (Department of Education, Training and EmploymentSPARQ-ed – University of Queensland Diamantina InstituteUniversity of Queensland, Australia.Page 4http://www.di.uq.edu.au/SPARQ-edph. 61 7 3443 6920 fax. 61 7 3443 6966Email: sparqed@uq.edu.au
Electron Microscopy – uses a focused beam of electrons instead of light. Electron microscopypermits a much higher magnification of specimens than light microscopy and is useful inobtaining detailed information about sub-cellular structures. Electron microscopy requiresextensive processing and so can only be performed on fixed specimens. Transmission electronmicroscopy provides a cross section of a specimen, while scanning electron microscopy gives athree-dimensional image of the surface of a specimen.Fluorescence Microscopy – uses fluorescent materials to indicate structures in a specimen.Fluorescence occurs when light of one wavelength “excites” a material and causes it to emitlight of a different wavelength. Most fluorescent materials give off visible light after excitationby ultraviolet light. Structures may be naturally fluorescent (autofluorescence) or they may belabeled with a compound which is fluorescent (eg. DAPI is a dye which binds onto DNA. TheDNA and nuclei of cells stained with DAPI emit a blue light under ultraviolet light).Immunofluorescence – antibodies are proteins made by the immune system which bind ontospecific parts of proteins. Antibodies can be raised against any protein in the cell. If theseantibodies are attached to a fluorescent tag, the tag will only show up where that antibodyattached (ie. where the target protein is found in the cell). Immunofluorescence allows veryspecific targeting of cellular structures.RNA Interference – RNA interference uses short sequences of RNA which are complementary to themRNA which carries to instructions to translate proteins from the DNA to the ribosomes. Theinterfering RNA binds to the target sequence, preventing it from being translated. As a result, carefulselection of interfering RNA can be used to silence a particular gene. This allows researchers to studywhat role a protein plays in a cell, by observing what happens when that protein is absent.Timelapse Microscopy – many cellular processes (eg. mitosis) occur over a period of time which is notpractical for direct observation. Imaging cells over a period of time (eg. a photograph is taken every 20minutes for 24 hours) allows us to combine these images in a “movie” which compresses a long timeperiod into a shorter one.The WorkshopWhilst the nucleus and some other organelles are easily visualized using brightfield microscopy, most othersub-cellular structures cannot be differentiated or even seen using conventional microscopes. Therefore thesestructures and biomolecules must be demonstrated using more specific techniques such asimmunofluorescence and fluorescence microscopy.In this project you will be provided with a coverslip containing cancer cells grown in culture. You will thenexpose the cells to antibodies directed against proteins found in subcellular structures, allowing these primaryantibodies to bind to those structures. You will then treat the cells with secondary antibodies directed againstthe primary antibodies. These secondary antibodies have been conjugated to a fluorescent dye, effectivelyattaching a fluorescent label to the structure you are interested in seeing. You will then use a fluorescencemicroscope to image the cells, taking images of each colour channel and then combining the images to createa merged image which demonstrates all structures. 2014 State of Queensland (Department of Education, Training and EmploymentSPARQ-ed – University of Queensland Diamantina InstituteUniversity of Queensland, Australia.Page 5http://www.di.uq.edu.au/SPARQ-edph. 61 7 3443 6920 fax. 61 7 3443 6966Email: sparqed@uq.edu.au
Getting StartedBefore you begin, make sure that you are familiar with the relevant theory behind the techniques we will beperforming. This manual contains several appendices which will provide you with this information. Make sureyou read this information before proceeding.Appendix A : DNAAppendix B : ProteinsAppendix A : Using a MicropipetteAppendix B : Glossary of TermsInformation on other cell and molecular biology concepts and techniques are provided at the SPARQ-edwebsite at : http://www.di.uq.edu.au/sparqedservicesTheoretical Basis of the ProjectTissue Culture and Cancer Cell LinesWhen researchers want to obtain sufficient quantities of organisms to use in their studies, they may growthem up in a vat of broth containing all of the nutrients they need to survive. This is called culturing theorganisms, and for many simple, free-living organisms such as bacteria or fungi, the process is relativelystraightforward. These organisms often require very little in the way of specialised nutrients and so long as theculture conditions are suitable, they will undergo almost constant division, increasing their populations at anexponential rate.If cells from a multi-cellular organism like a human are to be used in research, the culture methods are notnearly so simple. Firstly, cells from multi-cellular organisms have differentiated to such an extent that they canno longer survive without the complex systems of nutrients and stimuli provided by the other cells in the body.As a result, growing human cells in culture requires the use of growth media containing a complex mixture ofbasic nutrients and specific growth factors provided by the inclusion of serum (the liquid component of clottedblood). In addition, body cells spend most of their time carrying out the functions which allow them to playtheir roles within the body. This means that they are not constantly dividing - in many cases they must bestimulated to enter mitosis, and in some cases do not divide at all. As a result, human cells in culture oftenproliferate very slowly.One solution to this problem is the use of cancer cells. One of the hallmarks of cancer is the loss of regulationof the cell cycle, leading to cells constantly passing through division cycles. This results in hyperproliferation ofthe cells, which in the body leads to the growth of tissue masses known as tumours. Cancer cellhyperproliferation means that they can be grown in culture for many generations, with further cultures able tobe sub-cultured from the original – they are effectively immortal. This has led to the production of cancer celllines, each of which was derived from a tumour recovered from an individual with cancer.Most types of cancer have numerous cell lines which researchers can use to study the cancer in question. Eachof these cell lines have particular characteristics dependent on the cancer from which they were derived,allowing researchers to select the line which most closely matches the situation they are working on.Unfortunately, being cancer cells, the cells contain errors and abnormalities which mean that they do notoften behave as normal cells do, making them inappropriate models for normal cells, or even for cancers of 2014 State of Queensland (Department of Education, Training and EmploymentSPARQ-ed – University of Queensland Diamantina InstituteUniversity of Queensland, Australia.Page 6http://www.di.uq.edu.au/SPARQ-edph. 61 7 3443 6920 fax. 61 7 3443 6966Email: sparqed@uq.edu.au
other parts of the body. In addition, the same errors which remove the cell cycle controls in cancer cells mayalso remove mechanisms which regulate damage to the DNA, allowing mutations to accumulate in the cellswhich make them even more distinct from the cells from which they were derivedHeLa cells are possibly the best known of all cancer celllines. They were originally recovered from a cervical cancerremoved from a patient named Henrietta Lacks in 1951.The cancer resulted from an infection by a strain of humanpapillomavirus (HPV18), and so the genome of these cellsalso contains the genome for this strain of HPV. HeLa wasthe first cell line to be successfully and continuouslycultured in vitro and have been used widely around theworld since the physician who first subcultured them madethem and the techniques used to grow them freelyavailable to scientists around the world. HeLa cells haveplayed an important role in many important medicaldiscoveries, including the development of the Poliovaccine.Henrietta LacksThe patient fromwhom the HeLacell line was derivedwas Henrietta Lacks,who was living inBaltimore in the USat the time of herdiagnosis. While thecontribution that the HeLa cell line madeis well known, it is an interesting case ofmedical ethics, as permission for the useof her tissue was never sought. Moreinformation about the life of Ms Lacksand the issues around the use of her cellscan be found in the book The ImmortalLife of Henrietta Lacks by Rebecca Skloot.In this workshop, you will be using a genetically modifiedstrain of HeLa cells used by the Cell Cycle Research Groupat the University of Queensland Diamantina Institute.These cells have a modified gene for one of the histoneproteins in the nucleus (H2), where the gene includes agene derived from a jellyfish (the gene for greenfluorescent protein, or GFP). The histones are a family of proteins around which DNA wraps and which assist inthe packaging of DNA and help to regulate the expression of genes. When the modified gene is expressed, theprotein produced includes a GFP “tag” on the end which glows green when exposed to blue light. This hasproduced a cell line in which the nucleus fluoresces green, which means that this green signal can be used inplace of a DNA dye such as DAPI. This fluorescence is produced in living cells, allowing them to be used tostudy the function and appearance of the nucleus at different stages of the cell cycle through live cell imaging.Preparation of Cells for ImmunofluorescenceUsing routine brightfield microscopy, a reasonable amount of detail in the cell can be determined. Largestructures such as the nucleus (and its nucleoli) and vacuoles are easily distinguished inside the cell with evenfairly rudimentary microscopes. Some other structures can also be demonstrated with the careful use of dyesand stains which give a colour based on chemical reactions with cellular components, and this forms the basisof cytochemistry (on individual cells) and histochemistry (on thin sections of tissue). However, even thesemethods are not nearly specific enough to properly demonstrate the wide variety of subcellular structures andmaterials. Immunofluorescence is a technique which uses the highly specific binding of antibodies to theirtarget antigens as a way of demonstrating materials and structures inside cells.Before any immunofluorescence procedures can be carried out on the cells, they must be properly prepared.For this workshop, the cells have been grown on a coverslip placed on the bottom of a culture plate. When anew culture is needed, a suspension of cells is prepared with the cells floating around inside the cell culturemedium. The number of cells per unit volume (generally per millilitre of medium) can be calculated using cellcounters and a specific number of cells added to a dish containing culture medium by adding the correctvolume of cell suspension (eg. if the density of cells in the suspension is 100,000 cells / mL and you needed 2014 State of Queensland (Department of Education, Training and EmploymentSPARQ-ed – University of Queensland Diamantina InstituteUniversity of Queensland, Australia.Page 7http://www.di.uq.edu.au/SPARQ-edph. 61 7 3443 6920 fax. 61 7 3443 6966Email: sparqed@uq.edu.au
100,000 cells to start a culture, you would add 1mL of suspension). The exact seeding density depends on thereproductive rate of the cells you are using and how dense you want the culture to be in the time you have.When the cells are added to the culture dish, they sink to the bottom and attach through proteins embeddedin the cell membrane. Cells that have attached to the bottom of the dish take on the appearance of a fried egg,with the nucleus as the yolk and the cytoplasm as the white. Cells only release from the bottom of the flaskwhen they are in the process of dividing, becoming spherical as they carry out mitosis. For this workshop, thecells have been added to a 6-well culture plate. In each well, a number of coverslips were added to the wellsprior to the addition of the cells. When the cells settled to the bottom of the well, some landed on thecoverslip. This means that the cells can be easily removed from the well and treated while on the coverslip,and eventually the coverslip containing the cells can be mounted onto a microscope slide for observation. Thecoverslips have been previously treated with Poly-L-Lysine, a peptide which acts as a cellular “glue” and assistsin holding the cells (including mitotic cells which have rounded up) onto the coverslip.At several times during the course of the immunofluorescence procedures, cells will need to be washed. This isusually done by replacing the liquid in the well they are contained in with phosphate buffered isotonic saline, asolution of sodium chloride at 9g/L in a buffer which keeps the pH at a physiological level of around 7.2.Because the cells are stuck to the coverslip, the liquid can be gently removed and replaced with a solutionwhich does not put the cells under osmotic stress.Once cells die (which starts to occur once their nutrients and ideal growing conditions have been removed),they start to break down and lose their structure and integrity. A typical live cell taken through the processesneeded for immunofluorescence would have started the breakdown process by the time the techniques havebeen completed. Therefore the cells must be preserved prior to undergoing antibody treatment. This processis called fixation, and is the same technique used to preserve biological specimens in museums. Fixation relieson “freezing” the proteins in place using a chemical process rather than temperature. The tertiary structure ofproteins is maintained by weak hydrogen bonds between the side chains of the amino acids that make up theproteins. These hydrogen bonds are easily broken by increases in temperature or changes in pH, resulting in aloss of protein structure called denaturation. Formalin is a buffered solution of the gas formaldehyde whichforms permanent covalent crosslinks between protein chains and locks them into their tertiary structure,regardless of minor changes in temperature or pH, and thus preserves the structure of the cell. In thisinvestigation, you will use a type of formalin called paraformaldehyde to fix the cells, although other fixativeslike ice cold methanol can also be used.Once cells have been washed, fixed in paraformaldehyde and washed again, they must then be permeablised.The antibodies used for immunofluorescence are large protein molecules which cannot cross the cellmembrane, so the cell membrane must be removed. It is very important that the cells must be fixed prior topermeablisation, otherwise the cell will burst.To understand why it is important to fix the cells, it may help to imagine the cell as a dome tent filled withcooked spaghetti (with the nucleus as a beachball inside it, perhaps). The shape of the cell is maintained bystructural proteins such as Tubulin, represented in our analogy by the cooked spaghetti, while the whole shapeof the cell is maintained by the cell membrane, represented by the fabric of the tent itself. If you were to cutaway the tent fabric (removing the cell membrane through permeablisation), all of the spaghetti would fall outand spill onto the ground. Washing the cell would then have the effect of hosing the mess away.Fixing the cells would be like drying the spaghetti out. It would shrink slightly and revert back to its state priorto cooking, however it would be in the same position it was in when the drying occurred. The tent fabric (cellmembrane) could then be removed and you would be left with a mound of dried spaghetti in the same shapeas the tent, but without the tent around it (see Figure 1). Chemical fixation does make some changes to the 2014 State of Queensland (Department of Education, Training and EmploymentSPARQ-ed – University of Queensland Diamantina InstituteUniversity of Queensland, Australia.Page 8http://www.di.uq.edu.au/SPARQ-edph. 61 7 3443 6920 fax. 61 7 3443 6966Email: sparqed@uq.edu.au
proteins in the cell, however enough of the original structure is often left to retain some of the proteins’functions, including the ability of the proteins to bind antibodies and the fluorescence characteristics of greenfluorescent protein (both of which are vital for this investigation.Unfixed CellPermeablisationFixed CellPermeablisationFigure 1 - Fate of unfixed and fixed cells following permeablisationIn this workshop, cells will be permeablised using a detergent (Triton X100). Detergents work by solublisinglipids in water, and since the cell membrane is made up of lipid molecules, the Triton X100 in this methodremoves the lipids from the membrane, allowing large molecules such as antibodies to enter the cell and bindto proteins inside. At the same time as they are permeablised, the cells will be blocked. Blocking involves theaddition of a solution of proteins (usually bovine serum albumin, or BSA). Cells may contain proteins which canbind onto antibodies and other proteins non-specifically, rather than the antibodies specifically binding ontotheir target proteins. The BSA “soaks up” thesenon-specific binding sites, ensuring that the onlyVariable Regionsway the antibodies can bind is via their specifictargets. Once permeablised, blocked and washedonce more, the cells are ready to be treated withantibodies.Antibodies and ImmunofluorescenceAntibodies are large, complex proteins made byspecific immune cells in the body (the plasma cells,which are derived from populations of Blymphocytes). Antibodies are produced inresponse to the presence of an agent in the bodywhich the immune system recognizes as being notfrom the body. These agents are usually foreignmaterials, such as proteins on the surface ofmicrobes, although in some cases the immunesystem may target the body’s own proteins,causing autoimmune diseases such as Type IDiabetes and Rheumatoid Arthritis.Light ChainHeavy ChainFigure 2 Generalised Antibody Structure 2014 State of Queensland (Department of Education, Training and EmploymentSPARQ-ed – University of Queensland Diamantina InstituteUniversity of Queensland, Australia.Page 9http://www.di.uq.edu.au/SPARQ-edph. 61 7 3443 6920 fax. 61 7 3443 6966Email: sparqed@uq.edu.au
Antibodies are composed of four peptide chains (two light chains and two heavy chains) arranged like a capitalletter “Y” (see Figure 2). Most of an antibody’s structure is constant between antibodies (with some variationbetween different classes), however at the ends of the “arms” of the Y is a small, highly variable region madeup of a region of the light and heavy chains. It is changes in the shape of this region which determines thedifference between antibodies. The shape of this variable region is such that it fits around and binds veryspecifically to a region on the material it is raised against. The area which it binds to is called the epitope, whilethe material where the epitope is found is called the antigen. A different antibody is made in response to eachepitope on each antigen, with the variable regions confirming exactly to each epitope. This means thatantibodies are made which bind very specifically to particular epitopes on antigens. This specificity is so highthat two antibodies can be generated which can tell the difference between two versions of the same protein,one which has a phosphate group attached and one which does not. This high level of specificity makesantibodies extremely useful in detecting particular proteins, and is used in techniques such as diagnosis ofdisease agents in the blood, western blotting, immunohistochemistry, and, of course, immunofluorescence.Antibodies are made in animals, or in cell lines derived from those animals. To generate an antibody against aparticular protein, samples of that protein are injected into a test animal. A better antibody response isgenerated if the target protein comes from a different species to the animal used to raise the antibody as thehost animal would recognize the protein as being foreign. For example, if you are interested in usingantibodies to detect a human protein, you would use a non-human animal such as a mouse, rabbit or goat toraise the antibody. Once the animal starts making the antibody, it can be recovered from the animal’s bloodand purified for use. In some cases the lines of plasma cells which are making the antibody can be isolated andfused with a cancer cell, making a hybridoma cell line which has the immortality of the cancer cells and theantibody-generating properties of the plasma cells. This means that the antibodies can be produced in culturewithout the need for using further animals.The technique you will be using is indirect immunofluorescence. Direct immunofluorescence involveschemically combining a fluorescent dye directly to the primary antibody raised against the protein you areinterested in. The antibody, with its fluorescent label binds, onto the target protein in the cell, essentiallycolouring the target protein with the label (see Figure 3). Direct immunofluorescence is not often used now, asit would involve making a range of differently labeled antibodies for every protein studied in laboratories. Inaddition, the sensitivity of the technique is low, as there is only one label for each binding site.With indirect immunofluorescence, the primary antibody is left unlabelled. It still binds to the target protein,however it must be itself demonstrat
Cell biology is the study of cells and how they function, from the subcellular processes which keep them functioning, to the way that cells interact with other cells. Whilst molecular biology concentrates largely on the molecules of life (largely the nucleic acids and proteins), cell